T.C.
DĐCLE ÜNĐVERSĐTESĐ TIP FAKÜLTESĐ
ĐÇ HASTALIKLARI ANABĐLĐM DALI
AKUT LENFOBLASTĐK LÖSEMĐLĐ HASTALARDA
TĐOPÜRĐN S-METĐLTRANSFERAZ GEN
MUTASYONLARININ
BELĐRLENMESĐ
UZMANLIK TEZĐ Dr. Erhan ÖZENÇ
TEZ DANIŞMANI Prof. Dr. M.Orhan AYYILDIZ
1 TEŞEKKÜR
Uzmanlık eğitimim boyunca bilgi ve tecrübelerinden faydalandığım değerli hocam Đç Hastalıkları A.B.D. Başkanımız Prof. Dr. M. Emin YILMAZ’a, bilimselliği ile her zaman yanımızda olan Prof. Dr. Ekrem MÜFTÜOĞLU’ na, hiçbir zaman desteklerini esirgemeyen Prof. Dr. Vedat GÖRAL’a ve yetişmemde çok büyük emeği olan diğer değerli hocalarım; Prof. Dr. Abdurrahman IŞIKDOĞAN, Prof. Dr. Kendal YALÇIN, Doç. Dr. Alpaslan TUZCU, Doç. Dr. Ali Kemal KADĐROĞLU, Doç. Dr. Muhsin KAYA, Yrd. Doç. Dr. Hasan KAYABAŞI, Yrd. Doç. Dr. M. Ali KAPLAN, Yrd. Doç. Dr. Ali Đnal, Uz. Dr. Coşkun Beyaz, Uz. Dr. Yaşar YILDIRIM, Uz. Dr. Feyzullah Uçmak, Uz. Dr. Mehmet Küçüköner ve Uz. Dr. Remzi Beştaş’a teşekkürlerimi sunuyorum.
Bu çalısmanın her aşamasına bilgi ve tecrübesiyle katkıda bulanan, eğitimim ve tez çalısmalarım boyunca bana yol gösteren danışman hocam Prof. Dr. M. Orhan AYYILDIZ’a,
Bugüne kadar benim için hiçbir fedakarlıktan kaçınmayan, her zaman her koşulda bana destek olan sevgili eşim Ezra ÖZENÇ’e ve kıymetli aileme teşekkürü bir borç bilirim.
Sevgilerimle…
2 ĐÇĐNDEKĐLER Sayfa TEŞEKKÜR………...1 ĐÇĐNDEKĐLER………..….2 KISALTMALAR DĐZĐNĐ……….…..4 TABLOLAR VE ŞEKĐLLER DĐZĐNĐ……….………...6 ÖZET………..7 ABSTRACT ………..………...………....9 1. GĐRĐŞ………11 2. GENEL BĐLGĐLER………..…13 2.1. AKUT LÖSEMĐLER………13
2.1.1. Akut Löseminin Tanımı………...……13
2.1.2. Löseminin Tarihçesi……….…13 2.1.3. Löseminin Epidemiyolojisi………..14 2.1.4. Löseminin Patogenezi………..…14 2.1.5. Löseminin Etiyolojisi………...16 2.1.5.1. Çevresel Etkenler……….…16 2.1.5.2. Kalıtsal Etkenler……….…….17
2.1.6. Akut L ösemilerin Sınıflandırılması………..…..18
2.2. AKUT LENFOBLASTĐK LÖSEMĐ………....18
2.2.1. Klinik Bulgular………...…….20 2.2.2. Laboratuvar Bulguları………..…22 2.2.3. Tanı……….……..23 2.2.4. Ayırıcı Tanı……….…..24 2.2.5.Morfolojik Sınıflama………….………24 2.3.6. Sitokimyasal Boyama…………...25 2.2.7. Đmmünofenotiplendirme……….………..26
2.2.8. ALL’de Prognostik Faktörler…….………..…….27
2.2.9. Tedavi……….……….………..29
3
Sayfa
2.3.1. TPMT Transkripsiyonu………34
2.3.2. TPMT Geni………...34
2.3.3. Yaygın Mutant TPMT Alelleri………...34
2.3.3.1. TPMT *2………...37 2.3.3.2. TPMT *3B………...37 2.3.3.3. TPMT *3C………....……37 2.3.3.4. TPMT3A………...37 2.3.4. TPMT Genotip-Fenotip Đlişkisi………..…….38 2.4. TĐOPÜRĐN METABOLĐZMASI……….…39
2.4.1. Merkaptopürin, Tioguanin ve Azatiopürinin Etki Mekanizması………….40
2.4.2. Toksisite.………..…...43 2.4.3. Tedavi………..…….43 3. GEREÇ ve YÖNTEM……….……….45 3.1. Örnek Toplama….………45 3.2. Yöntem……….……45 4. BULGULAR………47 5. TARTIŞMA ve SONUÇ……….………..…49 6. KAYNAKLAR……….………54
4
KISALTMALAR DĐZĐNĐ
TPMT: Tiopürin S-metiltransferaz ALL: Akut Lenfoblastik Lösemi AML: Akut Myeloblastik Lösemi
CALLA: Common Acute Leukemia Leucosit Antigen CD: Cluster of Differanciation
EBV: Epstein-Barr Virüsü FAB: French- American- British HTLV: Đnsan T Hücreli Lösemi Virüsü LDH: Laktat Dehidrogenaz
HSM: Hepatosplenomegali MSS: Merkezi Sinir Sistemi SAM: S-Adenozilmetiyonin TGN: Tioguanin Nükleotidleri SNP: Tek Nükleotid Polimorfizmi adoMet: S-adenozil-L-metiyonin AdoHcy: S-adenozil-L-homosistein XO: Ksantin Oksidaz
HGPRT: Hipoksantin Guanin Fosforiboziltransferaz IMPDH: Đnozin Monofosfat Dehidrogenaz
GMPS: Guanin monofosfat sentaz
PCR: Polimeraz Zincir Reaksiyonu (Polymerase Chain Reaction) BOS: Beyin Omurilik Sıvısı
6-MeMP: 6Metil Merkaptopürin MPO: Miyeloperoksidaz SBB: Sudan Black
CAE: Choloroacetate Esterase
A-EST: Alpha Napthyl Acetate Esterase B-EST: Alpha Napthyl Butyrate Esterase PAS: Periodic Acide Schiff
5
TdT: Terminal Deoxynucleotidyl Transferaz Ig: Đmmünoglobülin
6
TABLOLAR DĐZĐNĐ
Sayfa
Tablo 2.1. ALL’ li çocuk ve yetişkinlerde klinik özellikler………...19
Tablo 2.2. ALL’li çocuk ve yetişkinlerde laboratuvar özellikleri………..23
Tablo 2.3. ALL’de FAB sınıflaması………..25
Tablo 2.4. Akut lenfoblastik lösemilerde sitokimya………...……26
Tablo 2.5. ALL ve immünofenotiplendirme………..27
Tablo 2.6. All olgularında prognostik faktörler……….28
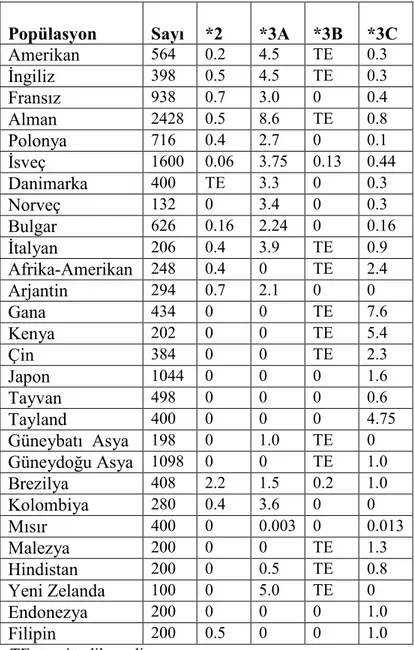
Tablo 2.7. Değişik popülasyonlarda TPMT allel sıklığı………..36
Tablo 2.8. Tiopürin S-Metiltransferaz allelleri………..38
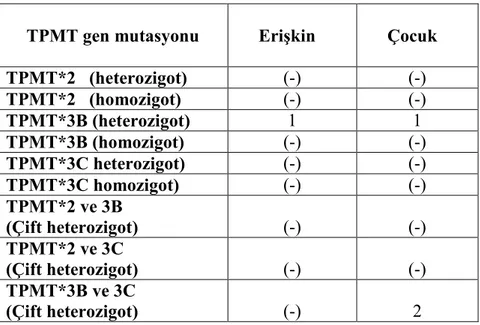
Tablo 2.9. ALL’li hastalar ve saptanan TPMT gen mutasyonları………...….48
ŞEKĐLLER DĐZĐNĐ Sayfa Şekil 2.1. Klinikte kullanılan tiopürin ilaçların kimyasal yapısı………..34
Şekil 2.2. TPMT ile S- adenozil L- homosisteinin kristal yapısı……….34
Şekil 2.3. Đnsan wild-tip TPMT (TPMT*1) geni ve yaygın mutant TPMT allelleri…..35
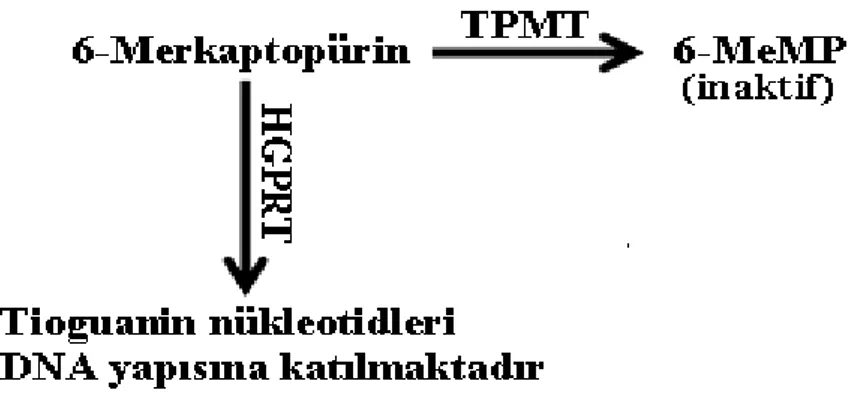
Şekil 2.4. 6-MP’nin aktif metabolitleri olan TGN’ye HGPRT tarafından dönüştürülmesi………39
7 ÖZET
AKUT LENFOBLASTĐK LÖSEMĐLĐ HASTALARDA TĐOPÜRĐN S-METĐLTRANSFERAZ GEN MUTASYONLARININ
BELĐRLENMESĐ
Bu çalışma, çocukluk çağı ve erişkin Akut Lenfoblastik Lösemili, tedavide 6-merkaptopürin kullanan hastalarda Tiopürin S-metiltransferaz gen mutasyonlarının araştırılması amacıyla yapılmıştır. Çalışmaya Temmuz 2007 - Temmuz 2010 tarihleri arasında Dicle Üniversitesi Tıp Fakültesi Erişkin ve Çocuk Hematoloji Bilim Dalında ALL tanısı konan ve tedaviye devam eden hastalar dahil edildi. 22’si erişkin, 33’ü çocuk olmak üzere toplamda 55 hasta çalışmaya alındı.
Tüm hastalara moleküler hematoloji labaratuvarında Real Time PCR yöntemiyle 3 farklı TPMT gen mutasyonu (TPMT*2, TPMT*3B ve TPMT*3C) bakıldı. Hastaların tümünde TPMT *2 gen mutasyonu (homozigot ve heterozigot) negatif olarak saptandı. Hastaların 2’sinde TPMT*3B gen mutasyonu heterozigot olarak bulundu (%3,6). 2 hastada TPMT*3B ve TPMT*3C çift heterozigot olarak saptandı (%3,6). Homozigot mutant bireylere rastlanmadı.
Erişkin hastaların 5’inde tedavi sırasında lökopeni gelişmişti. Bu hastalardan 4’ünde TPMT polimorfizmlerine rastlanmadı ancak 6-merkaptopürin dozu lökopeni nedeniyle azaltılmıştı (%25 oranında). Diğer 1 erişkin hastada TPMT*3B heterozigottu ve 6-merkaptopürin dozu lökopeni nedeniyle %50 oranında azaltılmıştı ve bir kez lökopeni ve enfeksiyon nedeniyle tedavisine ara verildiği görüldü. Tedaviye devam eden çocuk hastaların 30’unda lökopeni saptandı. Yine bunlardan TPMT*3B ve TPMT*3C çift heterozigot olan 2 çocuk hastanın belirgin lökopeni ve enfeksiyon nedeniyle zaman zaman tedavileri kesilmiş olarak saptandı. TPMT*3B heterozigot olan 1 çocuk hastada da lökopeni ve enfeksiyon nedeniyle tedaviye ara verildiği görüldü. Geriye kalan 27 çocuk hastada TPMT polimorfizmleri saptanmamış olup lökopeni nedeniyle doz azaltımı yapılmıştı (%25-50 oranında). Doz azaltımından yada tedavinin kesilmesinden sonra tüm hastalarda lökopenide düzelme saptandı. TPMT polimorfizmleri saptanmayan 5 çocuk hastada da hepatotoksisite bulgularına rastlandı.
TPMT polimorfizmi olan hastalarda, tedavide merkaptopürinin standart dozlarda verilmesi ağır toksisiteye (başlıca yan etki lökopeni) yol açabilmektedir. Bu nedenle
6-8
merkaptopürin kullanımı sonrası belirgin lökopeni gelişen hastalarda, TPMT mutasyonlarının analiz edilmesi oldukça önemlidir. Böylece ilaç dozlarının bireysel olarak ayarlanması tedavi etkinliğini maksimuma çıkarmakta, yan etkileri, muhtemel mortaliteyi ve tedavi harcamalarını da minumuma indirmektedir. TPMT mutasyonları ile ilgili yöremizdeki ilk çalışmalardan olan bu çalışmamız, ileride yapılacak olan kapsamlı populasyon çalışmalarınada zemin oluşturmuştur.
9 ABSTRACT
DETERMINATION OF THIOPURINE S-METHYLTRANSFERASE GENE MUTATION IN THE PATIENTSWITH ACUTE LYMPHOBLASTIC
LEUKEMIA
This study was carried out in order to investigate the Thiopurine S-Methyltransferase gene mutation in the patients with childhood an adult acute lymphoblastic leukemia using 6-Mercaptopurin for treatment. Patients who were diagnosed as ALL and received treatment in Dicle University Faculty of Medicine Departments of Pediatric and Adult Hematology between July 2007 and 2010 were enrolled to this study. Totally 55 patients were included, 22 of them were adults and 33 were children.
Three different TPMT gene mutation (TPMT*2,TPMT*3B and TPMT*3C) were tested in all patients in the molecular hematology laboratory with the Real Time PCR method. TPMT*2 gene mutation (heterozygosity and homozygote) was detected as negative in all patients. In 2 of the patients TPMT*3B gene mutation was detected as heterozygote (3,6%). It was detected as dual heterozygosity (TPMT*3B and TPMT*3C) in 2 of the patients (3,6%). No homozygote mutant individual was detected.
In 5 of the adult patients, leukopenia developed during the treatment period. In 4 of these patients, no TPMT polymorphism was detected but 6-Mercaptopurin dose was decreased due to the leukopenia (the rate of 25%). In other adult patient, TPMT*3B heterozygosity was detected and 6-Mercaptopurin dose was decreased at the rate of 50% due to the leukopenia. It was observed that because of the leukopenia and the infections the treatment was paused once. Leukopenia was detected in 30 of the children patients who continued the treatment. Also it was observed that the treatment of 2 of them, being TPMT*3B and TPMT*3C dual heterozygote, was quitted from time to time due to leukopenia and infection. It was observed that because of the leukopenia and the infections the treatment was paused in one child patient who was detected TPMT*3B heterozygote too. In remaining 27 children patients no TMPT polymorphisms were detected and leukopenia led to a decrease in
10
the dose (between 25%- 50%). After the decreases in the dose and pauses in the treatment, recovery was observed in the leukopenia. In five children patients that TMPT polymorphisms were not detected, hepatotoxicity symptoms were observed.
In the patients with TMPT polymorphisms, Standard dose of mercaptopurin in the treatment can cause severe toxicity (the main adverse effect is leukopenia). For this reason, in the patients developing clear leukopenia after the use of 6-mercaptopurin, analyzing the TMPT mutation is highly important. Thus, individual dose adjustment maximises the efficiency of treatment and minimises the adverse effects, possible mortality and the costs of treatment. This study which is one of the the first studies related to TPMT mutations in our region forms a basis for the future extensive population studies.
11 1.GĐRĐŞ
Akut lenfoblastik lösemi (ALL) kemik iliğinin klonal, malign hastalığıdır. ALL’deki malign hücreler gelişimlerin erken safasında duraklayan lenfoid öncül hücreleridir ve çoğalıp kemik iliğindeki normal hematopoeitik hücrelerin yerini alarak normal kan hücrelerin üretimini önlerler. Bunun sonucunda değişik boyutlarda anemi, trombositopeni ve nötropeni gelişebilmektedir (1,2).
Özellikle etkili ilaçların yanında destek tedavi yöntemlerinin de gelişmesi, merkezi sinir sistemi korumasının etkin yapılması, risk gruplarının daha doğru değerlendirilmesi ve hemetopoetik kök hücre naklindeki gelişmeler ile ALL hergün daha az korkulan bir hastalık haline gelmekte ve klonal eradikasyon ve kür olasılığı artmaktadır (3).
Akut lenfoblastik lösemilerde tedavi supportif bakım, kemoterapi, kemik iliği nakli gibi tedavi yaklaşımlarından oluşmaktadır. Kemoterapi esas olarak üç aşamadan oluşmaktadır (remisyon indüksiyon, konsolidasyon ve idame tedavileri). Đdame tedavisi konsolidasyonu takiben hastalığın nüks etmesini yani rezidü blastların öldürülmesi, normal kemik iliği hematopoetik progenitörlerin korunması amacı ile uygulanır. Đdame tedavisi süresi 2-3 yıl devam etmekte ve genellikle de haftalık metotreksat (20 mg/m²) ve günlük 6-merkaptopürin (yetişkinlerde 60 mg/m², çocuklarda 50 mg/m²) tedavisini içermektedir (4,5,6).
Tiopürin S-metiltransferaz tiopürin grubu ilaçların (azatiopürin, 6-merkaptopürin ve tioguanin) metabolizmasında rol oynayan bir enzimdir. Tiopürin grubu ilaçlar inaktiftir. Tiopürinlerin immünosupresif ve sitotoksik etkilerini göstermesi için tioguanin nükleotidlerine (TGN) metabolize edilmesi gerekmektedir. TPMT, bu ilaçları inaktive etmek için bunların S-metilasyonunu katalizler ve toksik olmayan metabolitler oluşturur. Sonuçta TPMT, intrasellüler sitotoksik TGN’lere dönüşümü önlemektedir. TPMT eksikliği olan hastalarda aktif TGN’lerin birikmesi sonucunda, ağır ve yaşamı tehdit eden hematolojik toksisite oluşabilmektedir (7,8,9,10).
TPMT aktivitesi trimodal dağılım göstermektedir. Rastgele seçilmiş büyük popülasyonlarda bireylerin %89’u yüksek TPMT aktivitesine sahip wild-tip, %11’i orta aktivite sahip heterozigot ve yaklaşık olarak 1/300 birey çok düşük veya hiç TPMT aktivitesi bulunmayan homozigot mutant bireylerden oluştuğu gösterilmiştir. Bu mutant
12
genlerden TPMT*2, TPMT*3A, TPMT*3B ve TPMT*3C düşük TPMT aktivitesinin %80-95’inden sorumlu olduğu tespit edilmiştir (10,11,12,13).
TPMT enzimini kodlayan genlerdeki polimorfizmler enzim aktivitesini değiştirmekte ve bireyler arasında enzimin farklı aktivitede çalışmasına neden olmaktadır. TPMT polimorfizmi olan hastalarda, tedavide merkaptopürinin standart dozlarda verilmesi ağır toksisiteye (başlıca yan etki lökopeni) yol açmaktadır.Bu nedenle seçilmiş olgularda tedaviden önce TPMT mutasyonlarının analiz edilmesi oldukça önemlidir. Böylece ilaç dozlarının bireysel olarak ayarlanması tedavi etkinliğini maksimuma çıkarmakta, yan etkileri ve tedavi harcamalarınıda da minumuma indirmektedir.
13
2. GENEL BĐLGĐLER
2.1. AKUT LÖSEMĐ
2.1.1. Akut Löseminin Tanımı
Akut lösemiler kemik iliğindeki kan hücrelerinin maturasyon ve farklılaşma özelliklerini kaybederek kontrolsüz çoğalması ile meydana gelen hastalıklardır (3). Çok fazla çoğalan fakat farklılaşmayan lökositler, başta kemik iliği olmak üzere karaciğer, lenf bezleri, dalak, deri, testis ve merkezi sinir sistemi (MSS) gibi organ ve sistemleri işgal edebilir. Bu hücrelerin kemik iliğindeki normal hematopetik hücrelerin yerini alması ve yeterince hücre üretilmesine rağmen tam olarak olgunlaşıp farklılaşamaması nedeniyle işlev gören hücre sayısı azalarak anemi, trombositopeni ve nötropeni oluşmaktadır. Bu tam olarak farklılaşmamış hücreler ‘blast’ olarak adlandırılmaktadır. Bu hücreler normal fonksiyonlarını yerine getirme yeteneğinden yoksundur. Lösemilerin kendi içindeki alt sınıflamalarında ve fazlara ayrımında bu blastların normal hücrelere oranı ve yapısı dikkate alınmaktadır. Etkili ilaç ve destek tedavileri sayesinde akut lösemiler kür sağlanan bir hastalık haline gelmiştir (14).
2.1.2. Lösemini Tarihçesi
Literatürde 1827’de Velpeau tarafından lösemi ile ilgili ilk vaka bildiriminden bu yana, lösemi gerek klinik gerekse genetik incelemelerde ilgi duyulan bir neoplazm olmuştur. Velpeau tarafından tespit edilen hastada; ateş, güçsüzlük, karın şişliği ve idrar yolları taşlarından kaynaklanan ağrılar şikayet edilmiş ve hastaneye başvurusundan kısa bir süre sonra hasta kaybedilmiştir. Hastanın otopsisinde çok büyük dalak ve karaciğerin yanı sıra, kanın kırmızı şarap renginde ve kıvamının artmış olduğu adeta lapa kıvamında olduğu bildirilmiştir . Bu tarihten 1845 yılına kadar lösemi çok iyi tanımlanamamıştır. Bu tarihlerde Virchow ve Benett ayrı ayrı olgularının otopsisinde, kanın beyaz rengine dikkat çekmiş ve 2 yıl sonra literatüre lösemi terimi kazandırılmıştır. Bu yüksek lökosit sayısına atfen beyaz kan terimi (weisses blut) kullanılmış, daha sonralarda ise Yunanca kökenli olarak beyaz anlamına gelen “leukos” ve kan anlamına gelen “haima” sözcüklerinden leukemia terimi türetilerek kullanılmaya başlanmıştır. 1913’lere gelindiğinde lösemi,
14
kronik lenfositik, kronik miyeloid, akut lenfositik, miyeloblastik, monositik, eritrolösemi olarak sınıflanıyordu. Literatürde bildirilen kür sağlanmış ilk lösemi olgusu 1930’da arsenik oksit, ışınlama ve iki kardeşten transfüzyonlarla hayatta kalan bir yetişkin hastadır. Belki de araştırmacı farkında olmasa da tarihte ilk miyeloablasyon ve periferik kandan kök hücre nakli yapılmıştı. Özel boyama teknikleri, elektron mikroskopi, kromozom analizleri, immünfenotipleme ve moleküler genotip belirleme gibi yöntemler geliştikçe lösemiler daha iyi anlaşılmıştır (15,16).
2.1.3. Löseminin Epidemiyolojisi
Akut Lenfoblastik Lösemi (ALL), çocukluk çağında en sık görülen malignite olup Amerika Birleşik Devletleri’nde 1-14 yaş grubu çocuklarda tanı alan kanserlerin %25-30’unu oluşturduğu bildirilmektedir. Ülkemizde bildirilen bir çalışmada çocukluk çağı maligniteleri içinde ALL sıklığı %34.9’dur. Çocukluk çağı akut lösemilerinin %80’i ALL olarak karşımıza çıkmaktadır. Erişkinlerdeki akut lösemilerin ise %20’sini oluşturur. Gelişmiş ülkelerde akut lösemilerin % 83’ünü ALL, % 17sini AML (Akut myeloblastik lösemi) oluşturmakta olup, insidansta bölgesel farklılıklar görülebilir. Ülkemizde kanser istatistikleri henüz yeterince sağlıklı olmamakla birlikte, ALL (çocuklarda) sıklık açısından yine birinci sırada yer almaktadır. Görülme sıklığı en fazla 3-5 yaş arasındadır ve daha sonra sıklık giderek azalarak yaşamın üçüncü dekatında yeniden artar. ALL’nin erken çocukluk çağında sık görülmesinin etiyolojik önemi açık değildir. Ancak gebelikteki olaylar ve immün sistemin gelişmesi ile ilişkili olduğuna dair görüşler vardır. ALL erkeklerde kızlardan daha sık görülür. Erkek/Kadın oranı 1,4/1,0’dır. T hücreli ALL’de ise bu erkek predominansı daha belirgindir (4:1). Cins prognoz açısından önemli bir gösterge olup kızlardaki sürvi oranı daha yüksektir (17,18).
2.1.4. Löseminin Patogenezi
Akut lösemilerin kesin nedeni tam olarak bilinmemektedir. Patogenezde tek bir mutasyondan daha çok, ardışık birkaç mutasyon sonrası oluşan mutant hücrenin çoğalması sorumlu tutulmaktadır. ALL’de sık görülen genetik bozukluklar konusunda yapılan araştırmalar sayesinde bugün hastalığın patogenez ve prognozu ile ilgili çok detaylı bilgilere ulaşılmıştır (19,20).
15
Tüm kan hücrelerinin kemik iliğinde tek bir kök hücresinden geliştiğine inanılmaktadır. Bu hücreler bütün hücre türlerini verdiği için ‘pluripotent kök hücresi’ olarak isimlendirilir. Pluripotent hücreler çoğalarak tek tür kan hücresini oluşturan ana hücreleri verirler. Bu hücrelerden bir kısmı ‘lenfoid’ diğer bir kısmı ise ‘myeloid’ hücre serisine farklılaşır. Myeloid hücreler kemik iliğinde gelişerek eritrositler, granülositler, monositler ve megakaryositlere dönüşürler. Gelişimin erken döneminde lenfoid hücreler kemik iliğinden lenf düğümleri, dalak ve timusa göç ederek lenfositlere farklılaşırlar. Yani hematopoez kök hücrelerinden gelişen ve farklılaştıkça dönüşme özellikleri azalan eş zamanlı ve devamlı olarak çeşitli hücrelerin üretilmesi ve bu hücrelerin farklılaşması işlemidir. Lösemi, kök hücreden spesifik kan hücrelerinin oluşumuna kadar herhangi bir farklılaşma aşamasında gözlenebilir (16).
Lösemik dönüşüm temelde bir mutasyon sonucu oluşmaktadır. Mutasyona uğrayan hücrelerin çoğu organizma tarafından çeşitli mekanizmalarla ortadan kaldırılmasına rağmen az sayıda hücre aşırı çoğalma özelliği kazanabilmektedir. Đşte bu hücreler “klonojenik lösemik hücreler” olarak adlandırılmaktadır (16).
Normal hematopoezde görev alan 12. kromozomdaki TEL geni ile 21. kromozom üzerinde bulunan AML1 genlerinin birleşmesi çocukluk çağı lösemilerinde %35’ e kadar varan oranda bildirilmiştir. Yenidoğanların kordon kanından yapılan bir örneklemede bu gen birleşim oranı % 1 bulunmuştur. Bu yaygınlık 15 yaşına kadar TEL-AML1 (+) lösemi tanısı alacak çocukların yaklasık yüz katı fazladır. Bu durumda prelösemik klonun hastalığa dönüşme oranı düşük olduğuna göre hastalığın oluşumu için prenatal olaya eşlik edecek bir veya daha fazla olaya ihtiyaç var demektir.
Çocuklar ve yetişkinlerde bazı farklılıklar gösterse de ALL’yi indükleyen genetik mekanizmalar benzerdir. Bu mekanizmalar;
• proto-onkogenlerin ekspresyonunda bozukluklar,
• kinaz enzimlerini aktifleştiren ve trankripsiyon faktörlerini etkileyen gen füzyonlarına yol açan kromozomal translokasyonlar,
• hiperdiploidi
olarak özetlenebilir. Sonuçta bu genetik değişiklikler hematopoetik kök hücrelerin lösemik transformasyonuna yol açar. Böylece hücre yenilenmesinde kontrol ortadan kalkar, normal proliferasyonu sağlayan kontroller etkisizleşir, olgunlaşmada duraksama olur ve apoptoz sinyallerine direnç gelişir.
16 2.1.5. Löseminin Etiyolojisi
Lösemilerin oluş sebebi tam olarak bilinmemekle birlikte meydana gelişinden tek bir faktörün sorumlu olmadığı da bir gerçektir. Muhtemelen çeşitli etkenler hastalığın oluşumunu hazırlamaktadır. Lösemi oluşumuna götüren etmenleri 2 grup altında inceleyebiliriz;
1. Çevresel (dış) etkenler 2. Bireysel (kalıtsal) etkenler
2.1.5.1. Çevresel Etkenler
Fiziksel ve kimyasal ajanlar ile virüsleri içeren çevresel etkenlerin karsinojenik etkisinin olduğu bilinmektedir. Bu karsinojenlerin kromozomlar üzerinde hasarlar yaparak kanser oluşturabildikleri bildirilmiştir. Karsinojenlerin kanser oluşturma riskleri; karsinojenlerin dozuna, etki süresine ve kişinin gen duyarlılığına bağlanmaktadır.
2.1.5.1.a. Radyasyon: Japonyaya ikinci dünya savaşı sırasında atılan atom bombalarından sonra lösemi sıklığı, özellikle 5-7 kat daha sık bulunmuştur. Radyasyonun dozu ile lösemi gelişimi arasında doğrudan bir alaka vardır. Hodgkin lenfoma veya ankilozan spondilit nedeniyle radyoterapi alan hastalarda akut lösemi riskinin arttığı bildirilmiştir. Özellikle radyasyon, hücrede öncül nükleotidlerin yapısında değişikliğe neden olmaktadır.
2.1.5.1.b. Benzen ve diğer kimyasal maddeler: Kimya, plastik, deri, boya ve lastik sanayi çalışanlarında akut lösemi riski, benzen veya petrol ürünleriyle temas nedeniyle yüksektir. Tütün kullanımı, petrol ve ürünleri ile sık ve doğrudan temas, etilen oksit, tarım ve böcek ilaçları kanser yapıcı olarak bilinmektedir.
2.1.5.1.c. Đlaçlar: Fenilbutazon, arsenik, etilen oksit ve kloramfenikol kullanımının akut lösemi riski taşıdığı bilinmektedir. Ancak asıl lökomojenik olanlar ise kanser ilaçlarıdır. Alkilleyici ilaçların (melfalan, klorambusil ve siklofosfamit) kullanımını izleyen 4. yılında lösemi riskinin belirgin arttığı bilinmektedir. Sekonder
17
lösemilerde kromozom anomalileri, özellikle delesyonlar sıktır. Özellikle etoposid kullanımı sonrasında sekonder lösemi gelişimi riski yüksektir.
2.1.5.1.d. Virüsler: Günümüzde en çok viral etyoloji üzerinde durulmaktadır. Kanser etyolojisinde rol oynadığı düşünülen virüsler; Retrovirüsler, EBV (Burkit Lenfoma ve ALL L3 morfolojik tip), HTLV-1 (Erişkin T-hücreli lösemi), HTLV-2 (Hairy cell lösemi)’dir.
2.1.5.2. Kalıtsal Etkenler
Genetik faktörlerin akut lösemilerin etyolojisinde, özellikle ALL’de, önemli rol oynadıkları ileri sürülmektedir. Özellikle familyal lösemi olgularının tahmin edilenden daha sık olması genetik faktörün lösemi etiyolojisinde rol oynadığı lehinde bir delildir.
2.1.5.2.a. Tek yumurta ikizleri: Genetik eğilimi destekleyen diğer bir unsur da monozigot ikizlerde, bir kardeşte lösemi görüldüğü zaman diğer kardeşte de lösemi görülme insidansının yüksek oluşudur. Çift yumurta ikizlerinde bu oranın düşüşü genetik yapının da olayla ilişkili olduğunu desteklemektedir.
2.1.5.2.b. Konjenital hastalıklar: Doğumsal kromozomal anomalileri bulunan olgularda malignite oranı sağlıklı bireylere göre yüksektir. Bu hastalıklar arasında en sıklıkla Down (21 trizomisi), Klinefelter (XXY ve varyantları), Patau (kromozom 13 trizomi) gibi sendromlarda görülür. Down sendromu olanlarda lösemi sıklığı (özellikle AML M7) normal populasyona göre 20-23 kat daha fazladır. Otozomal resesif kalıtım gösteren Ataksi telenjiektazi, Kseroderma pigmentozum, Fanconi anemisi, Bloom sendromu gibi durumlarda, çeşitli tip kanser vakalarında önemli artışlar gözlemlenmiştir.
2.1.5.2.c. Đmmün yetmezlikler: Doğuştan veya sonradan meydana gelen immün yetmezliklerde lösemiler daha sık görülür. Son yıllarda organ nakli nedeniyle uzun süre immünsupresif tedavi alan hastalarda sıklığı artmıştır. Splenektomi yapılmış Hodgkin hastalarında da akut lösemi riskinin arttığı düşünülmektedir.
2.1.5.2.d. Hematolojik hastalıklara sekonder: MDS veya miyeloproliferatif hastalıkların akut lösemiye dönüştüğü son derece iyi bilinmektedir (3).
18 2.1.6. Akut Lösemilerin Sınıflandırılması
Lösemiler, köken aldıkları hücre grubuna, semptomlarına, ortaya çıkış ve ilerleme hızlarına ayrıca klinik seyirlerine göre gruplara ayrılırlar. Lösemiler ortaya çıkış hızlarına göre öncelikle akut ve kronik olmak üzere 2 ana gruba ayrılırlar. Akut lösemiler de köken aldıkları hücre grubuna göre sınıflandırılırlar:
1. Akut Myeloblastik Lösemi (AML) 2. Akut Lenfoblastik Lösemi (ALL)
Her bir lösemi tipi de kendi içinde gelişiminin herhangi basamağında ya da evresinde olduğuna göre subtiplere ayrılırlar.
2.2. AKUT LENFOBLASTĐK LÖSEMĐ
Akut lenfoblastik lösemi (ALL) kemik iliğinin klonal, malign hastalığıdır. ALL’deki malign hücreler gelişimlerin erken safasında duraklayan lenfoid öncül hücreleridir ve çoğalıp kemik iliğindeki normal hematopoeitik hücrelerin yerini alarak normal kan hücrelerin üretimini önlerler. Bu duraksama çoğu zaman kromozomal translokasyonlarına ikincil anormal gen ekspresyonundan dolayı meydana gelmektedir. Bunun sonucunda değişik boyutlarda anemi, trombositopeni ve nötropeni gelişebilir. Lenfoblastlar karaciğer, dalak ve lenf nodları gibi kemik iliği dışındaki organlarda da çoğalabilirler.
Esas olarak, Akut lenfoblastik lösemi diğer malign lenfoid hastalıklardan, immünofenotiplendirme yöntemi ile B veya T hücreli olarak ayırt edilebilir. Malign lenfoid klonu berlilemekte yardımcı diğer yöntemler arasında ise immünokimyasal, sitokimyasal ve sitogenetik belirteçler gelmektedir.
ALL hastaları 2 ana mekanizmaya ikincil gelişen semptomlara bağlı sağlık merkezine başvurmaktadırlar:
1. Kemik iliği veya diğer organlarının lösemik hücreler ile direkt olarak infiltre olmasına bağlı semptomlar,
2. Normal kemik iliği elemenların üretimindeki azalmaya bağlı semptomlar. Kemik iliği infiltrasyonu sıklıkla kemik ağrılara neden olmaktadır. Bu ağrı çok şiddetli olabilidiği gibi, atipik prezentasyonlara neden olabilmektedir. Daha nadir olarak
19
(%10-20) splenomegaliye sekonder olarak hastalar sol üst kadranda dolgunluk ve erken doyma hissi ile başvurabilirler. Diğer yandan, özellikle T-hücreli ALL olguları büyük mediastinal kitleye bağlı bası semptomları ile başvurabilirler. Lenfoblastların aşırı sayılarda periferik dolaşıma katılmaları ile lökostaz belirtileri (solunum sıkınıtısı, zihin durumunda değişiklik) ile başvurabilirler ancak bu tablo AML hastalarındaki kadar sık değildir ve hücre sayımının mililitrede birkaç yüz bine ulaşana dek genelde gelişmez. Normal kemik iliği elemanların tükenmesine bağlı anemi (solukluk, çabuk yorulma, baş dönmesi, çarpıntı, efor dispnesi), nötropeni (artmış infeksiyon riski) ve trombositopeni gelişebilmektedir. ALL hastalarında aksi ispat edilene kadar infeksiyon en sık ateş nedeni olsa da, ateş primer hastalığın kendisine bağlı olarak ortaya çıkabildiği akılda tutulmalıdır (Tablo 2.1).
Tablo 2.1. ALL’ li çocuk ve yetişkinlerde klinik özellikler (16).
ÖZELLĐK ÇOCUK(%) YETĐŞKĐN(%)
Yaş <1 1-9 10-19 20-39 40-59 >60 3 77 20 - - - - - - 55 36 9 Erkek cinsiyet 55 62 Semptomlar Ateş Yorgunluk Kanama
Kemik ve eklem ağrısı
57 50 43 25 33-56 ? 3 25 Lenfadenopati Yok Var (>3cm) 30 15 51 11 Hepatomegali Yok
Var (umblikusun altında)
34 17 65 ? Splenomegali Yok
Var (umblikusun altında)
41 17 56 ? Mediastinal kitle 8 15 MSS tutulumu 3 8 Testiküler lösemi 1 0.3
20 2.2.1. Klinik bulgular
ALL’nin klinik bulguları oldukça farklılık göstermektedir. Hastaların yaklaşık 2/3’ünde hastalığın başlangıcı hızlıdır, tüm belirtiler 4 haftadan daha kısa bir sürede ortaya çıkmıştır. Bir grup hastada ise şikâyet ve bulgular sinsice ilerler ve tanıdan önce aylarca devam edebilir. En sık şikâyetler genellikle kemik iliği yetmezliği veya ekstramedüller yayılıma bağlı ortaya çıkmaktadır. Đlk şikâyetler genelde özgül olmayan halsizlik, yorgunluk, kemik ağrısı ve iştahsızlıktır. Ateş hastaların yaklaşık %55-60’ında vardır ve oldukça sık bir bulgudur. Çoğu olguda ateşin kaynağı lösemidir ve indüksiyon tedavisinin başlamasından sonra ilk 72 saatte kaybolur. Hastalarda enfeksiyon insidansı ve prevalansı mutlak nötrofil sayısı ile ters orantılıdır ve bu sayı 500/mL ve özellikle 100m/L’nin altına indiğinde enfeksiyon riski artmaktadır. Bununla birlikte hastalar genelde nötropenik olduğundan ve var olan nötrofiller de fonksiyonel olarak anormal olabileceğinden tüm febril hastalar infeksiyon ayırt edilinceye kadar geniş spektrumlu antibiyotikler ile tedavi edilmelidir (20-21).
2.2.1.1. Kas iskelet sistemi bulguları: Özellikle küçük çocuklarda eklem ve kemik ağrıları sıktır. En sık alt ekstremiteler tutulur. Lösemik hücrelerin periostu tutmasından, kemik enfarktından ya da kemik iliği mesafesinin artan blast yükü ile genişlemesinden kaynaklanır. Belirgin kemik ağrıları olan hastaların çoğunda hematolojik parametrelerin normal değerde olması ve bazı hastalarda periferik kanda lösemik hücre bulunmayışı (alösemik lösemi) tanıyı geciktirmektedir. Bu hastalar artrit, artralji veya romatoid artrit tanısı ile izlenebilirler.
2.2.1.2. MSS bulguları: Lösemik hücreler hematojen yayılımla veya daha nadir olarak kafatası kemiklerin tutulumuna ikincil araknoid yüzeyine köprüleşen venler aracılığı yayılabilir. Hastalığın tanısı, seyri sırasında veya remisyonda iken gelişebilir. Tanı sırasında %5’den daha az olguda saptanır. MSS tutulumu özellikle matür B-ALL ve T-ALL’de daha sık görülür. Tanı için BOS sitosantrifüj sonrası May–Grünwald ve Giemsa ile boyanarak dikkatli bir şekilde değerlendirilmelidir. Tanı anında MSS tutulumu olan çocuklarda yaygın veya fokal nörolojik bulgular ortaya çıkabilir. Artmış intrakraniayal basınç bulguları, halsizlik, baş ağrısı, kusma, papilla ödemi görülebilir. Ayrıca kafa çifti sinirlerinin tutulumuna bağlı sıklıkla yüz felci görülebilir.
21
2.2.1.3. Genitoüriner sistem bulguları: Genellikle lenfatik obstruksiyona ikincil testiste ağrısız şişlik şeklinde ortaya çıkar. Tedavinin bitmesine rağmen testislerde büyüme devam ediyorsa veya testis relapsını kanıtlamak için biyopsi yapılabilir. Nadiren sakral köklerin tutulumu veya korpora kavernezum ve dorsal venlerin blastlar tarafından tutulması ile priapizm gelişir. T hücre veya olgun B hücreli lösemide ise böbrek tutulumu saptanabilir. Hematüri, hipertansiyon veya böbrek yetersizliği klinik olarak tespit edilir (22).
2.2.1.4. Gastrointestinal bulgular: Ağızda kandida infeksiyonu sıktır. Mukozal ülserasyonlar, peteşi ve ekimozlar görülebilir. Lenfadenopati, splenomegali ve hepatomegali gibi organomegaliler saptanabilir. GĐS kanaması olabilir. Trombositopeni, yaygın damar içi pıhtılaşması, lösemik hücre infiltrasyonu ve kandida gibi infeksiyonlar kanamaya yol açabilir. Tiflit, çekumda nekrotizan enterokolit gelişmesidir. Ağır nötropeni ve sepsis varlığında sağ alt kadran ağrısı, hassasiyet, abdominal distansiyon bulgularıyla belirti verir. Ultrasonografi ile barsak duvar kalınlığında artış saptanabilir.
2.2.1.5. Göz bulguları: Göz tutulumu çok dikkatli bir inceleme sonucunda yeni tanı almış ALL hastalarında saptanabilir. Trombositopenin neden olduğu retinal hemoraji özellikle lökosit sayısı yüksek hastalarda gözün lösemik tutulumuna neden olur (23).
2.2.1.6. Mediastinal ve Kardiyopulmoner bulgular: Otopsilerde kalp tutulumu gösterilmesine rağmen %5’ten daha az olguda klinik bulgu oluşur. Patolojik olarak miyokard ve perikardda blast saptanır. Lösemik infiltrasyon veya kanamaya bağlı oluşur. Ayrıca lösemilerin %5-10’unda tanı sırasında mediasten kitlesi saptanır ki, bunların çoğu T-hücreli lösemidir.
2.2.1.7. Deri bulguları: ALL’de oldukça nadirdir, AML’de daha sık saptanır. Deri altında nodül olabilir (leukemia cutis).Trombositopeniye bağlı kanama sonrası lösemik hücrelerin deride proliferasyonuna bağlı ortaya çıkabilir (22).
22 2.2.2. Laboratuvar bulguları
Anemi, anormal lökosit sayıları ve trombositopeni kemik iliğinin lösemik hücrelerce işgalini yansıtır (Tablo 2.2). Tanıda hastaların 1/3’inde 5.000/mm³yarısında ise 10.000/mm³’ün altındadır. Hiperlökositoz (>100.000/mm³) hastaların %10-15’inde görülür. Belirgin granülositopeni ise (<500/mm³) %40 oranında görülür. Periferik kan yaymada görülen hücrelerin büyük bir kısmı lökosit ve lenfoblasttır. Lenfoblastlar dar sitoplazmalıdır. Ancak lökopenili hastaların periferik yaymasında blastlar görülmeyebilir.
Trombosit sayısının düşüklüğü tanıda hastaların %92’sinde görülen bir bulgudur. Lösemide izole trombositopeni pek nadir olduğu için immün trombositopeniden ayırtetmek güç değildir. Trombosit sayısı 20.000/mm³değerleri gibi düşük dahi olsa, ateş veya infeksiyon gibi riskler yoksa ciddi kanama pek görülmez.
Anemi hastaların %75’inden fazlasında görülür, normokromik normositiktir, retikülosit sayısı normal veya düşüktür.
Kemik iliğinde genellikle %80-100 oranında blast vardır. Tanı için kemik iliğinde %20’den yüksek blast oranı gereklidir. Kemik iliğinde mutlaka histokimyasal, immünfenotipik ve sitogenetik inceleme yapılmalıdır.
Artmış lösemik hücre yükü pürin katabolizması yolu ile serum ürik asit düzeylerinde artışa sıklıkla neden olur. Serum LDH düzeyi de lösemik hücre yükünün göstergelerindendir ve genellikle kötü prognozla ilişkilidir.
Hiperkalsemi hastaların %0.5’inde görülür; lenfoblastların parathormon benzeri bir protein salınımına yol açmasından veya kemiğin lösemik infiltrasyonuna bağlı ortaya çıkmaktadır. Bu komplikasyon genellikle hidrasyon ve kemoterapinin başlaması ile kendiliğinden düzelir.
Lösemik infiltrasyona bağlı karaciğer fonksiyon bozukluğu tanı anında %10-20 oranında görülür, genelde hafiftir, önemli bir klinik veya prognostik değeri yoktur.
PA Akciğer grafilerinde mediastinal genişleme veya efüzyon görülebilir.
Beyin omurilik sıvısı (BOS) incelemesinde mikrolitrede 5 veya daha fazla blast olması veya kranial sinir tutulumu olması MSS lösemik tutulum olarak değerlendirilir. BOS kısa sürede sitosantrifüj sonrası incelenmelidir.
23
Tablo 2. 2. ALL’li çocuk ve yetişkinlerde laboratuvar özellikleri (16)
ÖZELLĐK Çocuk(%) Yetişkin(%)
Lökosit saysı <10.000 10.000-49.000 50.000-99.000 >100.000 50 31 9 10 41 31 12 16 Hemoglobin konsantrasyonu <8 8-10 >10 52 28 20 28 26 46 Platelet sayısı <50.000 50.000-100.000 >100.000 48 20 32 52 22 26 Kemik iliğinde blast
oranı(%) <90 >90 19 81 29 71 Kanda lösemik blast
Var Yok 84 16 92 8 2.2.3. Tanı
ALL tanısı fizik muayene, kan sayımı, periferik yayma, kemik iliği ve BOS’un sitolojik incelemesi ile konur. Kemik iliğindeki %20 veya daha yüksek oranda lenfoblast görülürse ALL tanısı kesinlik kazanır. Kemik iliğinde fibroz veya çok yoğun lösemik infiltrasyon aspirasyona engel olabilir. Bu hastalarda biyopsi gereklidir. Biyopsi yapılan dokunun imprint ile hazırlanan preparatları morfolojik tanı için boyanabilir. Kemik iliği nekrozu olan olgularda canlı doku alabilmek için aspirasyon işleminin defalarca tekrarlanması gerekebilir.
24 2.2.4. Ayırıcı Tanı
ALL’li hastalarda başlangıç belirtileri akut eklem romatizması ile karışabilir. Akut başlayan kanama belirtileri akla idiyopatik trombositopenik purpurayı getirebilir. AML veya ALL’de aplastik anemide olduğu gibi pansitopeni görülebilir. Aplastik anemide lenfadenopati, hepatosplenomegali, olması akut lösemileri veya lenfomayı düşündürür. Đnfeksiyoz mononükleoz ve diğer viral enfeksiyonlarda trombositopeni ve çevre kanında atipik lenfositlerin görülmesi ayırıcı tanıda yardımcı olur. Diğer lökomoid reaksiyonlar ve hepatosplenomegaliyle birlikte olan pansitopenili hastalarda akut lösemi ile ayırıcı tanı gerekebilir. Bu hastalarda kemik iliği incelenmesi çok önemlidir. Miyeloproliferatif hastalıkların blastik fazı ve miyelodisplastik sendromlu hastalar akut lösemi ayırıcı tansında problem oluşturabilirler. Bu hastalarda periferik yayma, kemik iliği incelemelerinin yanı sıra, anamnez, klinik bulgular, sitogenetik incelemeler yardımcıdır (3).
2.2.5. Morfolojik Sınıflama
ALL’yi sınıflandırmak, tedavi ve prognozu bu sınıflandırmaya göre belirleme çabaları 30 yılı aşkın bir süredir devam etmektedir. Üzerinde uzlaşmaya varılan ilk sınıflandırma 1976’da Bennett ve arkadaslarının yayınladığı Fransız, Amerikan ve Đngiliz Hematologlarının ortak çalısması olan FAB sınıflamasıdır (Tablo 2.3). Bu sistem klasik ışık mikroskopisi ile lenfoblastların morfoloji değerlendirmesini içerir. FAB sınıflamasına göre lösemik hücrelerin L1, L2 ve L3 olmak üzere 3 subtipi vardır.
L1 alt tipi daha çok çocuklarda görülmekte; açık mavi boyanan, monomorfik, yuvarlak çekirdekli, dar sitoplazmalı, küçük ve orta büyüklükte blastlar, homojen nükleer kromatin yapısı ve belirsiz nükleoluslar dikkat çekicidir.
L2 alt tipine erişkinlerde sıklıkla rastlanır. Blastlar büyük ve hetorojen, daha geniş sitoplazmalı ve değişken görünümlüdür. Nükleostoplazmik oranı daha düşüktür. Düzensiz kenarlı çekirdek yapısı ve belirgin nükleuslar görülmektedir.
L3 tip çok nadir görülür, hücreler koyu bazofilik sitoplazma ve sitoplazmik yağ içeren vakuoller ile karakterizedir. Bu grup Burkit lenfoma ile aynı morfolojidedir.
Morfolojik sınıflama giderek önemini kaybetmektedir. Bunlar yerine, monoklonal antikorların kullanıldığı yüzey belirleyici analizi, sitoplazmik belirleyiciler, kromozom yapısı ve yeniden gen düzenlenmelerinin analizine başvurulmaktadır. Bu
25
çalışmalar ALL’nin sürekli olarak tanımlanan yeni alt grupları ile büyük orandaki heterojenitesini ortaya koymuştur. Özellikle ALL’nin AML’den ayırımında ve kendi içinde alt tiplere ayırımında monoklonal antikorlardan yararlanılır (6).
Tablo 2.3. ALL’de FAB Sınıflaması (24).
Sitoloji L1 L2 L3
Hücre boyutu Küçük Büyük, heterojen Büyük, homojen Nükleer
kromatin
Homojen Değişken,
heterojen
Noktalı ve homojen Nükleus şekli Düz konturlu,
bazen çentikli
Đrregüler, sıklıkla çentikli
Düzgün konturlu, oval yuvarlak
Nükleolus Görülmez veya silik, küçük
Bir veya birden fazla,
sıklıkla belirgin
Belirgin, bir veya birden fazla, veziküler
Sitoplazma Dar Değişken,
sıklıkla büyük
Orta derecede büyük Sitoplazmik
bazofili
Hafif veya orta, nadiren koyu Değişken, bazen koyu Çok koyu Sitoplazmik vakuol
Değişken Değişken Sıklıkla belirgin
2.2.5.1. L1 tip ALL: Küçük ve homojen hücrelerden oluşur. Hücrelerin %25’i T lenfosittir. Nükleus membranı düzenlidir. En sık görülen kromozom anomalileri t(9;22), t(1;19), t(4;11), t(8;14), 6q ve 9p anormallikleridir.
2.2.5.2. L2 tip ALL: Hücre sitoplazması daha geniş olup, bir veya daha fazla belirgin nükleolus vardır. Hücrelerin %25’i T lenfosittir. Karşılaşılan kromozom anomalikleri t(9;22), t(4;11), 6q, +21, +8, i(17q), 7p-,11q-‘dir.
2.2.5.3. L3 tip ALL: Hücreler büyük ve homojendir. ALL’nin %5’inden fazlası bu tiptir. Pek çok hücre olgun B lenfositlerden köken alır. Kromozom anomalileri t(8;14), t(8;22), t(2;8), 1q-, +6, 6q-, 8q+ dir.
26 2.2.6. Sitokimyasal Boyama
Blastların bazı boyaları alıp almama veya az ya da çok almaları da lösemi tipini belirlemek için faydalı bir özelliktir (Tablo 2.4). Miyeloperoksidaz (MPO), Sudan Black (SBB), Periyodik-Aside-Schiff (PAS), Asid Fosfataz (AP), Nonspesifik Esteraz (NSE), Terminal Deoksinükleotil Transferaz (TdT), Demir (Fe) boyaları kullanılmaktadır. Bunların farklı kombinasyonlarda pozitif olması FAB sınıflamasını desteklemektedir (4,26).
Tablo 2.4. Akut lenfoblastik lösemilerde sitokimya
FAB MPO SBB AP CAE
A-EST B-EST PAS MGP Fe TdT L1 - -/np +* - +/z -/z +/- + - + L2 - -/np +* - -/z -/z +/- + - + L3 - - +* - - - +/- + - -
np: nadir pozitif, *: T-ALL’de unipolar pozitif, z: zayıf,
MPO:Miyeloperoksidaz, SBB: Sudan Black, CAE: Choloroacetate Esterase, A-EST:
Alpha Napthyl Acetate Esterase, B-EST: Alpha Napthyl Butyrate Esterase, PAS: Periodic Acide Schiff, MGP: Methyl Gren Pyronine, TdT: Terminal Deoxynucleotidyl Transferaz
2.2.7. Đmmünofenotiplendirme
ALL, hücre yüzeyi antijen ekspresyonuna dayanan birçok gruba ayrılabilir. En sık 5 şekil; erken pre-B-hücreli, pre B-hücreli, B-hücreli, T-hücreli ve nötr-hücreli (null-cell) ALL’dir (Tablo 2.5).
ALL’li olguların yaklaşık %60’ı hücre yüzeyinde ortak ALL antijenini (CALLA) eksprese eder. CALLA (CD10), bazı normal genç lenfositlerde ve diğer hematopoetik dokular dışındaki dokularda bulunan bir glikoproteindir. CALLA (+) ALL olgularının erken pre-B-hücre farklılaşma durumunu temsil ettiği düşünülmektedir.
CALLA (+) ALL’nin yaklaşık %20’sinin stoplazmik immünoglobulinleri vardır ve pre-B-hücreli ALL olarak adlandırılırlar.
B-hücreli ALL, hücre yüzeyinde immünoglobulin bulunması ile belirlenir ve ALL olgularının %5’inden azını oluştururur.
27
ALL olgularının yaklaşık %20’si hücre fenotipindedir ve normal genç T-hücrelerinde bulunan CD5, CD3, CD2 gibi antijenleri eksprese eder.
ALL olgularının yaklaşık %15’inde CALLA veya T-hücre belirteçleri yoktur ama bir B-hücre belirteci olan CD19 eksprese edilir, pro-B-ALL olarak adlandırılır.
ALL’li hastaların %25’inde lösemik hücreler myeloid belirteçleri de eksprese eder. Bu antijenlerin varlığının eskiden daha kötü prognoza sahip bir grubu tanımladığı düşünülürdü ama günümüzde agresif rejimlerle hastalık seyrinin benzer olduğu görülmektedir (27).
Tablo 2.5. ALL ve immünofenotiplendirme (16)
Subtip Tipik Markırlar Çocuk(%) Yetişkin(%)
Prekürsör B hücre Pre-pre-B Erken pre-B Pre-B CD19+, CD22+, CD79a+, cIg+-, HLADR+ CD10- CD10+ CD10+-, cIg+ 5 63 16 11 52 9
B hücreli CD19+, CD22+, CD79a+, cIg+ 3 4
T hücre kökenli Thücre Pre-T CD7+, cCD3+ CD2+, CD1+-, CD4+-, CD8+-, HLADR-, TdT+- CD2-, CD1-, CD4-, CD8-, HLADR+-, TdT+ 12 1 18 6
cCD3: Sitoplazmik CD3, cIg: Sitoplazmik immünoglobulin, TdT: Terminal deoksinükleotidil transferaz
2.2.8. ALL’de prognostik faktörler
ALL hastalarında prognostik kriterlerin klinik takipte büyük önemi vardır (Tablo 2.6). Bu faktörler:
28
1- Yaş: AML’de olduğu gibi önemli bir prognostik kriterdir. Çocuk yaş grubu ALL’lerde 1 yaş altı ve 10 yaş üstü olguların prognozu ara yaş grubuna göre daha kötüdür.
2- Cinsiyet: Kadınlarda erkeklere göre prognozun daha iyi olduğu düşünülür. Bu duruma erkeklerde görülen testiküler relapsın etkisi olabilir. Bu duruma rağmen sadece testis relapsı kötü prognozu izah etmemektedir.
3- Lökosit sayısı: Lökosit sayısının 10.000/ mm³’ün altında olmasının iyi, 50.000/ mm³’ün üstünde olmasının ise kötü prognoz göstergesi olduğu belirtilmektedir.
Tablo 2.6. All olgularında prognostik faktörler (3).
Đyi prognoz Kötü prognoz Lökosit sayısı <10.000/mm³ >50.000/ mm³
Yaş 2-10 yaş <1 yaş, >10 yaş
Cinsiyet Kadın Erkek
Irk Beyaz Siyah
LAP, HSM Yok Var
Mediastinal kitle Yok Var
MSS tutulumu Yok Var
FAB ALL-L1 ALL-L2, ALL-L3
Hemoglobin >10gr/dL <7g/dL
Trombosit >100.000/mm³ <30.000/mm³
Remisyon statüsü (28.gün sonuda)
Tam remisyon (+) Tam remisyon (-)
Serum Ig Normal Azalmış
Đmmünofenotipleme Erken Pre-B hücreli T hücreli, B hücreli
Sitogenetik işaretler Hiperploidi 6q- T(12-21) Psödoploidi t(9-22), t(8-14), t(4-11), t(14q+), t(1-19), p16, p16 kaybı, 9p21 delesyonu
Diğer Kötü performans durumu
Düşük albümin düzeyi Yüksek LDH düzeyi LAP: Lenfadenopati, HSM: Hepatosplenomegali, MSS: Merkezi sinir sistemi, Ig: Đmmünoglobülin
4- Đmmünofenotipik özellik: Olgun B hücre ve T hücreli ALL olgularında izlenen kötü prognozla birlikte immünofenotipik özelliklerin ALL’deki önemi belirginleşmiştir. Đlginç bir şekilde, çocukların aksine yetişkinlerde T hücre özelliği her
29
zaman kötü prognoz göstergesi değildir. Genel olarak erken prekürsör B hücreli ALL’nin iyi prognozlu olduğuna inanılır. Bu noktada önemli olan sitoplazmik Ig’dir. Yani yüzey Ig taşımayan ALL hastaları taşıyana göre daha iyi prognoza sahiptir.
5- Kromozomal bozukluklar: Yüksek hiperploidi (50’den fazla kromozom veya DNA indeksinin 1.16’dan büyük olması) ve kromozom 4 ve 10 trizomileri iyi prognoz göstergesidir. Kötü kromozom anormallikleri arasında hipoploidi (<45 kromozom), MLL genin düzenlenmesi (kromozom 11q23) ve philadelphia kromozomu ﴾t(9-22)﴿ bulunmaktadır. t(1-19) tarnslokasyonunun kötü prognostik özelliği intensif tedaviyle ortadan kaldırılabilir. Sonuç olarak hiperploidi, 6q- ve t(12-21) iyi prognoz göstergesidir. Psödoploidi, t(9-22), t(8-14), t(4-11), t(14q+), t(1-19), 9p21 delesyonu ve p16 delesyonunun ise kötü prognostik kriter olduğu kabul edilmektedir.
6- Đndüksiyon tedavisine hızlı cevap: Đndüksiyon tedavisine hızlı yanıt, uzun süreli sağ kalım üzerine olumlu etkisi olduğu bilinen önemli bir prognostik kriterdir. Tedavi sonrası cevap alınıyorsa bu iyi prognoz göstergesi sayılmalıdır. Remisyon için 2 veya daha fazla indüksiyon tedavisi alması gereken hastalarda prognozun ilk ay içinde yanıt alanlara göre belirgin derecede kötü olduğu bilinmektedir (3).
2.2.9. Tedavi
Özellikle etkili ilaçların yanı sıra destek tedavi yöntemlerinin de gelişmesi, merkezi sinir sistemi korumasının etkin yapılması, risk gruplarının daha doğru değerlendirilmesi ve hemetopoetik kök hücre naklindeki gelişmeler ile ALL hergün daha az korkulan bir hastalık haline gelmekte ve klonal eradikasyon ve kür olasılığı artmaktadır. Hala prognozu yetişkinlerde kötüdür. Akut lenfoblastik lösemilerde tedavi supportif bakım, kemoterapi, kemik iliği nakli gibi tedavi yaklaşımlarından oluşmaktadır. Kemoterapi esas olarak üç aşamadan oluşmaktadır (remisyon indüksiyon, konsolidasyon ve idame tedavileri). Kemik iliğindeki blast sayısının %5’ten daha az olması, klinik ve periferik kan değerlerinin (trombosit ve nötrofil sayıları) düzelmesi tam remisyon olarak tanımlanmaktadır. Hastalarda remisyon elde edildikten sonra hastalığın tekrarlamasına relaps veya nüks ismi verilir. Đdame tedavisinin sonlanmasını izleyen altıncı aydan sonra olan relapsa geç relaps, indüksiyon sonrası remisyonu takiben tedavi sırasında veya
30
idame tedavisinin sonlanmasını izleyen ilk altı aylık sürede herhangi bir anda oluşan relapsa erken relaps adı verilir. Refrakter veya direçli olgu ise remisyon indüksiyon tedavisi ile remisyona sokulamayan hastlara verilen isimdir. Bu durumda kök hücre nakli gibi daha agresif tedavi yaklaşımları uygulanmalıdır. Prekürsör B hücreli ve T hücreli ALL’de tedavinin süresi 2-3 yıl arasında devam eder. Özellikle philadelphia kromozom pozitifliği başta olmak üzere kötü prognostik kriterler içeren olgularda kemik iliği nakli uygulamaları önemlidir. Yetişkin olgularda tam remisyon oranları % 70-90, uzun süreli sağ kalım oranları ise sadece %20-50’dir. Genel olarak idame tedavi yaklaşımları yetişkin hastalarda çocuk yaş grubuna göre daha agresiftir (3).
2.2.9.1. Supportif Bakım: Lösemilerde supportif bakım genel olarak santral venöz kateter konulması, sitopenilerin replasmanı (trombosit ve eritrosit süspansiyonları), DIC profilaksisi ve tedavisi, tümör lizis sendromuna uygun medikal yaklaşım, hiperlökositozis halinde lökoferez ve/veya uygun tıbbi müdahale, enfeksiyona karşı profilaksi ve tedavi, hastaya ve ailesine psikososyal destek, erken ve geç yan etkilerin engellenmesi veya azaltılmasına yönelik yaklaşımlar şeklinde özetlenebilir (4,5,6).
2.2.9.2. Kemoterapi:
2.2.9.2.a. Remisyon Đndüksiyon: Hastaları klinik olarak remiyona sokmayı hedefleyen bir tedavidir. Bu tedavi aşamasındaki hedef lösemik blastların kemik iliğindeki oranının %5’in altına indirilmesidir. Remisyon indüksiyon tedavisinde bir çok farklı rejimde farklı ilaçlar kullanılmakla beraber vinkristin ve prednizon temel ilaçlardır. L-Asparaginaz ve antrasiklinlerde (daunorubisin, doksorubisin) genellikle yetişkin hastalarda tedaviye eklenmektedir. Bu ilaçların birlikte kullanılması remisyon oranını %75-90’a yükseltmektedir. Yüksek risk gruplarında dörtlü ilaç kullanımı daha çok tercih edilmektedir. Alkilleyici ilaçlar (siklofosfamit), epipodofilotoksinler (etoposid), antimetabolitler (metotreksat ve sitozin arabinozid) de remisyon indüksiyon tedavisinde kullanılabilir. Remisyon indüksiyon tedavisi ile remisyon cevabı alınamayan hasta sayısı genellikle %10’dan azdır. Bu tür olgular tedaviye dirençli sayılarak daha agresif tedavilere adaydırlar. Bu hastalarda sağ kalım oranları ve prognoz kötüdür.
31
2.2.9.2.b. Konsolidasyon: Hastalara remisyon indüksiyon tedavisi ile remisyona girdikten sonra remisyonun devamını sağlamak için konsolidasyon tedavileri uygulanır. Konsolidasyon tedavisi; daha yüksek dozda ve indüksiyondaki ilaçlarla çapraz direnç oluşturmayan ilaçlar kullanılarak yapılır. Daha dirençli olan blastların yok edilmesini amaçlar. Genellikle bu dönemde MSS tutulumuna yönelik profilaktik veya terapötik radyoterapi uygulanır (3,28).
2.2.9.2.c. Reindüksiyon: Bazı protokollerde kısa bir ara idame periyodunu takiben indüksiyon ve konsolidasyon karışımından ibaret olan reindüksiyon fazı uygulanmaktadır.
2.2.9.2.d. Đdame: Konsolidasyonu takiben hastalığın nüks etmesini yani rezidü blastların öldürülmesi, normal kemik iliği hematopoetik progenitörlerin korunması amacı ile uygulanır. Đdame tedavisi süresi 2-3 yıl devam etmekte ve genellikle de haftalık metotreksat (20 mg/m²) ve günlük 6-merkaptopürin (yetişkinlerde 60 mg/m², çocuklarda 50 mg/m²,) tedavisini içermektedir. Bazı protokollerde bu tedaviye vinkristin ve kortikosteroit de eklenmektedir.
2.2.9.3. Merkezi Sinir Sistemi Lösemisi Profilaksi ve Tedavisi: Lösemik blastların MSS’de sekestrasyonu ve daha sonra sistemik relaps oluşumuna sebep olmasından dolayı ayrı bir tedaviye ihtiyaç vardır. MSS tutulumu yok ise profilaktik, var ise terapötik amaçla tedavi verilmektedir. ALL’li hastaların %3’ünde tanı sırasında MSS tutulumu saptanabilir. MSS profilaksisi için yüksek doz metotreksat ve/veya Ara-C veya ĐT metotreksat ve 1800 cGy kranial radyoterapi uygulanmalıdır. MSS tutulumunda tedavi yaklaşımı ise üçlü ĐT tedaviye (metotreksat, Ara-C, hidrokortizon) ek olarak 2400 cGy kranial ve 1200-1500 cGy spinal radyoterapi uygulanmaktadır. Bu yaklaşımlarla önceleri %50 olan MSS relaps oranı %5’lere indirilmiştir. Ayrıca bu uygulamalar genel sürvi oranını da artırmıştır.
2.2.9.4. Testiküler Lösemi Tedavisi: Lösemide testis tutulumu testiküler büyümenin yanında testis biyopsisi yapılarak lösemik infiltrasyonun gösterilmesi ile kanıtlanmalıdır. Testis biyopsisi ile %33 oranında asemptomatik tutulum tespit edilmiş
32
olsa da rutin yapılması önerilmemektedir. Asemptomatik vakalarda sistemik kemoterapi dışında ilave bir müdahale yapılmaz. Semptomatik ve biyopsi ile ispatlanmış hastalarda ise kemoterapiye ek olarak 1200 cGy testiküler radyoterapi önerilmektedir. Đyi tedavi edilmemiş erkek çocuklarda testiküler lösemi %10 oranında geç relapslara neden olmaktadır.
2.2.9.5. Tedavi süresi: Đdeal tedavi süresi genellikle kesin olmamakla birlikte, idame tedavisi ile birlikte 2-5 yıl sürmektedir. 5 yıldan daha uzun tedavi önerilmemektedir. Bununla beraber, uygulanan tedavi rejimi, hastalığın risk grubu, tedavi yanıtı gibi bir çok faktör tedavi süresi üzerine etkili olduğundan, idame tedavi süreleri hakkında kesin öneriler yoktur. Ancak tedavi süresinin iki yıldan az olması genellikle tasvip edilmemektedir.
2.2.9.6. Philadelphia kromozom pozitif ALL’de tedavi: Philadelphia kromozomu ﴾t(9-22)﴿, yetişkin hastalarda yaklaşık % 40, çocuklarda ise %5 oranında pozitiftir. Uzun süreli sağ kalım oranları bu gruplarda oldukça düşüktür. Philadelphia kromozomu pozitif ALL hastalarında remisyon süresinin çok kısa olduğu bilindiğinden 1. remisyonda allojenik kök hücre nakli yapılmalıdır. Kronik myelositik lösemi tedavisinde kullanılan ve bir tirozin kinaz inhibitörü olan imatinib bu grup hastalarda kullanılabilmektedir.
2.2.9.7. Relaps Tedavisi: Yapılan çalışmalar çocukluk çağı ALL olgularının %20-30’unun, yetişkinlerde görülen ALL hastalarının ise %70-80’i remisyon indüksiyon tedavisi ile remisyona girmelerine rağmen takip sırasında relaps olduklarını göstermiştir. Relapsların %80 kadarı kemik iliği, %12-16’sı MSS, %8’i testis relapsı şeklindedir. Relaps ALL olgularında prognoz üzerinde belirleyici olabilecek bazı faktörler vardır. Bunlar arasında remisyonda kalma süreleri yani relaps olana kadar geçen zaman, relapsın yeri, immünofenotipik karakterler, yaş, relaps sırasındaki toplam lökosit sayısı gibi faktörler sayılabilir. Bir çok deneyimli merkez yukarıda sayılan faktörler arasında en çok erken relapsı önemsemektedir. Burada erken relaps olarak ilk iki yıl içinde ortaya çıkan relaps kastedilmektedir. Yapılan çalışmalar, ilk iki yıl içinde relaps olguların, geç relaps olanlara göre daha kötü prognoza sahip olduklarını göstermiştir. Relapsın
33
lokalizasyonuda önemlidir. Ekstramedüller relapsın (genellikle MSS), sistemik relapsa göre daha iyi prognozlu olduğu bilinmektedir. Hatta hem kemik iliği hemde ekstra medüller relaps olgularının sadece kemik iliği relapsına göre daha iyi prognozlu olduğuda bildirilmiştir. Ekstramedüller nüksler içinde ise testiküler relapsın, MSS relapsına göre daha iyi prognoza sahip olduğu düşünülmektedir. Relapslarda (özellikle tekrarlayan relapslarda) ilaçlara karşı direnç gelişimi tedaviye cevapta önemli rol oynar. Yeni tanı almış vakalarda sürvi oranları yüksek iken relaps yapmış hastalarda bu oranın oldukça düşük olduğu saptanmıştır. Relaps vakaları için üzerinde fikir birliği yapılan protokol ya da protokollar mevcut değildir. Genellikle hastanın önceden almış olduğu kemoterapi protokolünden daha yoğun bir protokol seçilmektedir (3,5).
2.3. TĐOPÜRĐN S-METĐL TRANSFERAZ ENZĐMĐ
Tiopürin S-metiltransferaz azatiopürin, 6-merkaptopürin ve tioguaninin metabolizmasında rol oynayan bir enzimdir. Enzim bir metil donörü olarak S-adenozilmetiyonin (SAM) kullanarak sülfür atomlarının metilasyonunu katalizlemektedir. TPMT, tiopürin ilaçlarını (Şekil 2.1) inaktive etmek için bunların S-metilasyonunu katalizleyen ve toksik olmayan metabolitler oluşturan sitoplazmik bir enzimdir (7,8,9,10). TPMT reaksiyonu metil donörü S-adenozil-L-metiyonin (adoMet) ile metil akseptörü olan iki substratı almakta ve S-adenozil-L-homosistein (AdoHcy) ile metillenmiş ürün oluşturmaktadır (Şekil 2.2). Bu enzimin katalizlediği reaksiyon aşağıda gösterilmiştir (29,30).
TPMT
S-adenozil-L-metiyonin + Tiopürin <==> S-adenozil-L-homosistein + Tiopürin S-metileter
Metil akseptörü olarak 6-merkaptopürin ve diğer tiopürinler çok iyi karakterize edilmesine rağmen, bu enzim için bugüne kadar doğal bir substrat tanımlanamamıştır. Pürin analogları olarak dizayn edilen bu ilaçlar, normal nükleotid homeostazda olduğu
34
gibi aynı metabolik yol ile metabolize edilerek normal DNA sentezini bozmak için hedeflenmiştir. Bu ilaçlar, hücrelerde sellüler DNA yapısına giren ve programlanmış hücre ölümünü tetikleyen 6-TGN’lere dönüştürülmektedir. TPMT ile katalizlenen S-metilasyon, tiopürinlerin biyoyararlanımını azaltmaktadır. Bu nedenle TPMT, intrasellüler sitotoksik 6-TGN’lere dönüşümü önlemektedir (7,33).
Şekil 2.1. Klinikte kullanılan Şekil 2.2. TPMT ile S- adenozil-
Tiopürin grubu ilaçların kimyasal yapısı (31). L-homosisteinin kristal yapısı (32).
2.3.1. TPMT’nin Transkripsiyonu
TPMT, tiopürin ilaçların S-metilasyonunu katalizleyerek inaktif ve toksik olmayan metabolitler oluşturan sitoplazmik bir enzimdir. Bu enzim için bugüne kadar doğal bir substrat tanımlanmamasına rağmen, TPMT transkriptleri akciğer, karaciğer, iskelet kasları, böbrek, lenfositler, platelletler, kırmızı ve beyaz kan hücrelerini içeren değişik doku tiplerinde eksprese edilmektedir. TPMT aktivitesi tüm dokularla korelasyon göstermekte olup, kolay elde edilebilmesinden dolayı kırmızı kan hücrelerinde ölçülmektedir (7,8,9,10,33).
2.3.2. TPMT Geni
TPMT geni 6p22.3 kromozomu üzerinde yerleşmiş olup, 9 intron ve 10 ekson tarafından kodlanmaktadır (Şekil 2.3). Bu genin kodladığı protein, 28 kDa moleküler
35
ağırlığında ve 245 amino asitten oluşmuştur. TPMT eksikliği otozomal kodominant kalıtım göstermektedir (7,8,10,33).
2.3.3. Yaygın Mutant TPMT Allelleri
Birçok çalışmada tiopürin ilaçlarının neden olduğu hematopoeitik toksisitesinin majör faktörün TPMT geninde inaktif enzime neden olan tek nükleotid polimorfizmlerinin (SNP) olduğu tanımlanmıştır. TPMT polimorfizmleri terapötik etkinlik ve merkaptopürinlerin toksisitesi ile ilişkilendirilmiştir. Bugüne kadar çalışılmış bütün büyük popülasyonlarda, TPMT aktivitesinin yüksek derecede değişken ve polimorfik olduğu gösterilmiştir. TPMT’nin trimodal kalıtım gösteren üç temel enzim fenotipi (düşük, orta ve yüksek) vardır. Popülasyonun %89’u wild-tip olarak adlandırılan normal/yüksek TPMT aktivitesine sahiptir. Kalan %11 ise TPMT mutant alleli için heterozigotluğu belirten orta enzim aktivitesine, her 300 kişiden 1 kişi TPMT için çift heterozigot veya homozigot olan düşük yada hiç tespit edilemeyen TPMT aktivitesine sahiptir. (11,12,13).
TPMT*1 olarak adlandıran wild-tip dışında 26 farklı mutant TPMT alleli literatürlerde tanımlanmıştır. Bütün bu allellerde TPMT aktivitesi düşük ve/veya homozigotlarda enzim aktivitesi tespit edilmemiştir. Bunlardan dört varyant (TPMT*2, TPMT*3A, TPMT*3B ve TPMT*3C) düşük TPMT aktivitesinin %80-95’inden sorumludur.
36
Şekil 2.3. Đnsan wild-tip TPMT (TPMT*1) geni ve yaygın mutant TPMT allelleri. Siyah kutular
eksonları, beyaz kutular ise translate edilmeyen bölge dizilimini kodlayan eksonları veya eksonların bir kısmını göstermektedir (9).
Beyaz ırkta, Afrika-Amerikalı ve Asya popülasyonlarında mutant allellerin yaklaşık %90’ı üç mutant allel (TPMT*2, TPMT*3A ve TPMT*3C) oluştururken, diğer mutant alleller sadece birkaç vakada tespit edilmiştir.
Tablo 2.7. Değişik popülasyonlarda TPMT allel sıklığı (46,47,48). Popülasyon Sayı *2 *3A *3B *3C
Amerikan 564 0.2 4.5 TE 0.3 Đngiliz 398 0.5 4.5 TE 0.3 Fransız 938 0.7 3.0 0 0.4 Alman 2428 0.5 8.6 TE 0.8 Polonya 716 0.4 2.7 0 0.1 Đsveç 1600 0.06 3.75 0.13 0.44 Danimarka 400 TE 3.3 0 0.3 Norveç 132 0 3.4 0 0.3 Bulgar 626 0.16 2.24 0 0.16 Đtalyan 206 0.4 3.9 TE 0.9 Afrika-Amerikan 248 0.4 0 TE 2.4 Arjantin 294 0.7 2.1 0 0 Gana 434 0 0 TE 7.6 Kenya 202 0 0 TE 5.4 Çin 384 0 0 TE 2.3 Japon 1044 0 0 0 1.6 Tayvan 498 0 0 0 0.6 Tayland 400 0 0 0 4.75 Güneybatı Asya 198 0 1.0 TE 0 Güneydoğu Asya 1098 0 0 TE 1.0 Brezilya 408 2.2 1.5 0.2 1.0 Kolombiya 280 0.4 3.6 0 0 Mısır 400 0 0.003 0 0.013 Malezya 200 0 0 TE 1.3 Hindistan 200 0 0.5 TE 0.8 Yeni Zelanda 100 0 5.0 TE 0 Endonezya 200 0 0 0 1.0 Filipin 200 0.5 0 0 1.0
37
Beyaz ırkta enzim aktivitesini azaltan en yaygın polimorfizmin TPMT*3A olduğu belirlenmiştir. TPMT*3A, beyaz ırkta %5 olarak tespit edilmiştir. TPMT*3C ise en yaygın olarak Afrika ve Asya popülasyonlarında bulunmakta ve düşük enzim aktivitesine neden olmaktadır. Asya ülkelerinde enzim eksikliğinin çok az olduğu tespit edilmiştir. Batı Asya’da, popülasyonunda baskın olarak TPMT*3A görülmüştür. Doğu Asya’da ise TPMT*3C baskındır (9,12,13,46,47,48).(Tablo 2.7).
TPMT*2, TPMT*3A, TPMT*3B ve TPMT*3C genotipinin tespit edilmesi durumunda TPMT aktivitesi tahmin edilebilir; tüm dört allelin heterozigot formu hastada orta aktiviteye, bu allellerin homozigot formunda ise hastada TPMT aktivitesi gözlenmez. Ek olarak, çift heterozigotlarda da (TPMT*2/3A, TPMT*2/3C, TPMT*3A/3C) hiç TPMT aktivitesi gözlenmemektedir.
Yapılmış olan birçok çalışmada TPMT eksikliği olan hastalarda tiopürinlerin konvansiyonel dozajı ile tedavi edilirse, ağır hematopoeitik toksisite gelişme riskinin çok yüksek olduğu bulunmuştur. TPMT lokusu heterozigot olan hastalarda da orta düzeyde toksisite riskinin olduğu gösterilmiştir. Buna zıt olarak enzim aktivitesi çok yüksek olan hastalarda, bu ilaçların standart dozu ile tedavilerinde yanıt alınmayabilir (7,33,34).
2.3.3.1. TPMT*2
TPMT*2 allelinde 80. rezidüde bulunan alanin prolin ile yer değiştirmiştir (Tablo 2.8). Bunun sonucunda TPMT aktivitesi yaklaşık 100 kat azalmakta ve çok düşük immünolojik protein düzeyine neden olmaktadır. TPMT*2 proteinin yarı ömrü 0.35 saate sahiptir ve ubikuitin yolu ile proteozomlarda degrade edilmektedir (35,36).
2.3.3.2. TPMT*3B
A154T (154. rezidüde bulunan alanin treonin ile yer değiştirmiştir) polimorfizmi (Tablo 2.8), wild-tip TMPT ile karşılaştırıldığında immünolojik protein düzeyinde azalmaya neden olmaktadır ve proteinin 24 saatten fazla olan yarı ömrünü 5.5 saate düşürmektedir. Bu da protein düzeyinin dört kat azalması ile sonuçlanmaktadır (35).
38 2.3.3.3. TPMT*3C
TPMT*3C allelinde (Y240C) 240. rezidüde bulunan tirozin sistein ile yer değiştirmiştir (Tablo 2.8). TPMT*3C, wild-tip proteinine göre immünolojik proteinin ve enzim aktivitesinin düzeyi daha düşük olarak saptanmıştır (35).
2.3.3.4. TPMT*3A
TPMT*3A alleli, A154T ve Y240C polimorfizmlerinin her ikisini bulundurmaktadır (Tablo 2.8). Bu allelde, protein düzeyinin ~400 kat azalmasına ve ölçülemeyecek enzim aktivitesine neden olmaktadır. TPMT*3A, wild-tip proteinden yaklaşık 72 kat daha az olan ~0.67 saat yarı ömrü ile hızlıca ubikuitin yoluyla proteozomal olarak degrade olmaktadır (35).
Tablo 2.8. Tiopürin S-Metiltransferaz Allelleri
TPMT Aleli Gen Mutasyonu Aminoasit Değişimi Aktivite (%) *1 Wild tip 100 *2 G238C Ala80→Pro 26 *3A G460A A719G Ala154→Thr Tyr240→Cys 1.6 *3B G460A Ala154→Thr 1.7 *3C A719G Tyr240→Cys 17 *3D G460A A719G G292T Ala154→Thr Tyr240→Cys Glu98stop *5 T146C Leu49→Ser 2.4 *6 A539T Exon 8’de transversiyon Tyr180→Phe 36 *7 T681G Exon 10’da transversiyon His227→Glu 98 *8 G644A Arg215→His 82 *10 G430C Gly144→arg 69 *11 G395A C132→Y 36 *12 C374T Ser125→Leu 41 *13 A83T Glu28→Val 61 *16 G488A Arg163→His *19 A365C Lys122→Thr