Neuroprotective strategies against calpain-mediated neurodegeneration
Tam metin
Şekil
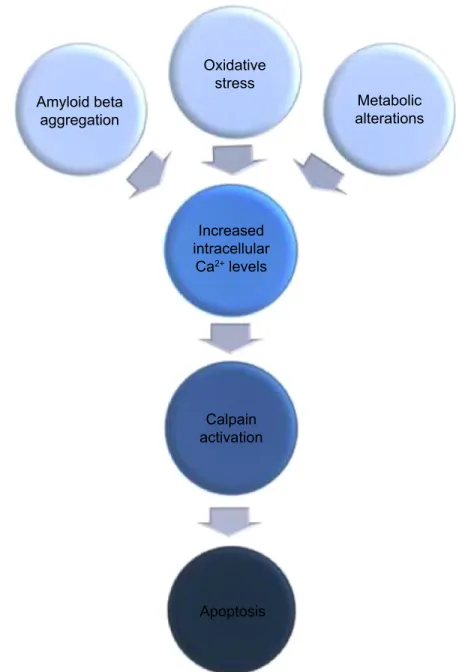
Benzer Belgeler
Bu araştırma kapsamında, bugüne kadar reklam ve müzik ilişkisi konusunda gerçekleştirilen marka ta nınırlığı, reklama karşı olumlu tutum, markaya karşı olumlu
Emzirme döneminde ilaç kullanımının bebek üzerinde olası advers etkileri ile ilgili endişeler nedeniyle an- nenin tedavisiz kalması, anne ve bebek açısından istenmeyen kalıcı
Sonuç olarak bu çalışmada vejeteryan diyet ve yüksek posa tüketiminin divertiküler hastalıkta hastaneye yatma veya hastalıktan kaynaklanan ölüm ile ilişkili olduğu
Yüksek sıcaklıktaki austenit fazın uzun süren dönüşümü sonucunda, şekil hafızalı alaşımlarda, termoelastik martensitin oluşması işlemi martensitik
Japonya'daki folklorcular, inceleme saha larının birbirine yakınlığından dolayı, kendi araştırm a alanlarinı Antropoloji, Sosyoloji ve ta rih gibi daha önce
Şu kadarım söyleyim : Faruk Nafiz şiire çok genç yaşta başlamış ve daha ilk şiirleri onu kuşağının en güçlü şairi olarak zamanın üstad- larına
Protestan kilisesinin öncülük ettiği, Amerika’daki küçük Ermeni kolonisinin ve Amerikan basınının da katıl dığı bu kampanyada, Türkiye en ağır
Sultan Mehmet devrinin bu büyük bes tekârı tarz ve edasındaki yenilik, nağme lerindeki yüksek ruh ve derin mâna ile klâsik Türk musikisinin başlı
