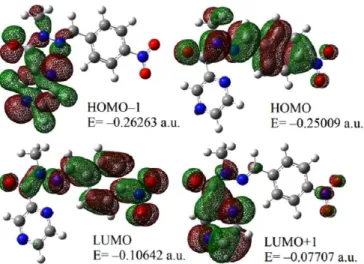
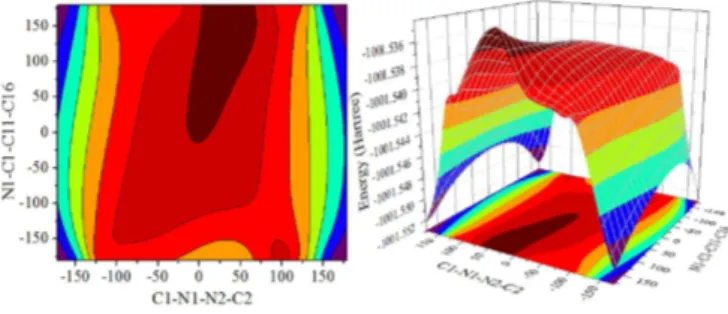
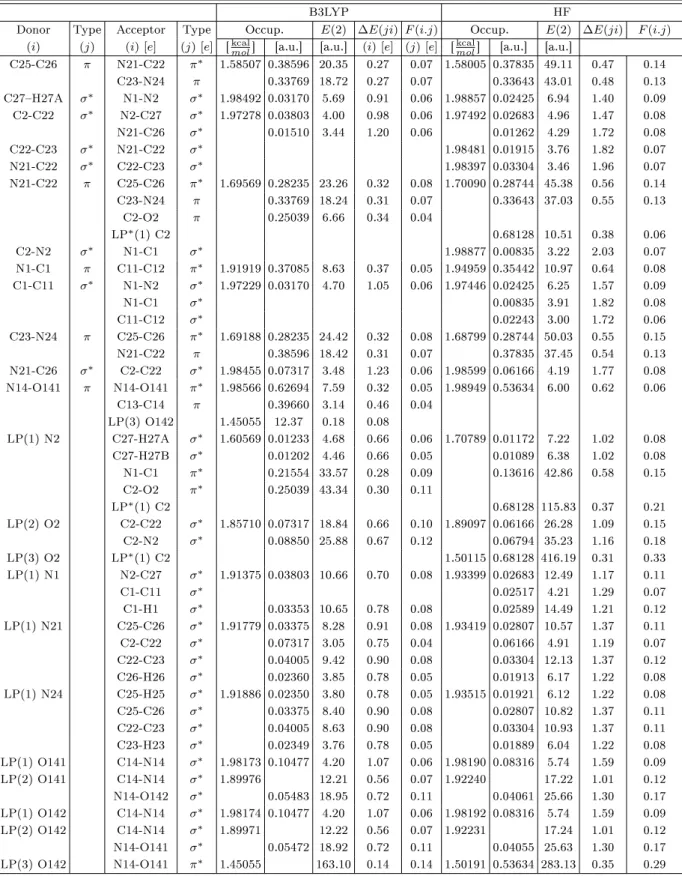
Theoretical Investigation of N-Methyl-N '-(4-nitrobenzylidene) pyrazine-2-carbohydrazide: Conformational Study, NBO Analysis, Molecular Structure and NMR Spectra
Tam metin
Şekil
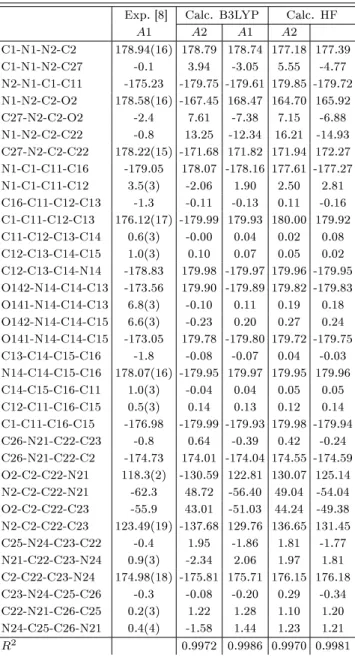
![Fig. 1. (a) The experimental geometric structure [8], (b) optimized molecular structure (with B3LYP/6-311++G(d,p) level) of the title compound.](https://thumb-eu.123doks.com/thumbv2/9libnet/3998687.54445/2.892.484.826.157.771/experimental-geometric-structure-optimized-molecular-structure-level-compound.webp)

Benzer Belgeler
This study argues that the hybrid quality of the party created by a diluted version of the Ke- malism and social democracy will benefit Kiliçdaroğlu. CHP will not be divided into
Çalışmamızda 15-15-15 gübresi; tomurcuk sayısını en çok etkileyen gübre olurken biyokütle artışını ise en çok Ozmokot (9 ay) gübresi etkilemiştir. iberica)‟nin
In this study, we proposed novel trimming approaches for AFOs such that the trimlines were performed on the dorsal side rather than lateral and medial sides to reduce the magnitude
DFT’nin, sıkıs¸tırmalı algılama alanındaki sonuc¸ların yarattı˘gı beklentiye, y¨uksek olasılıkla iyi performans garantileri sa˘glıyor olmasına ve bazı ¨ozel
In this article, we devised query processing strategies that use the result entries found in the result cache of a search engine to answer previously unseen user queries.. We
The aim of the study is to understand the effects of speech and speech intelligibility on computer based task performance in open-plan offices and examining if the performance
Kişileri hayatın getirdiği her türlü olumsuzluklara karşı koruması ve bundan daha önem lisi iç h u z u r ve asayişin sağlanarak, cem iyet hayatını ahenkli
1) Zaman ilişkili faydalar: Lojistik köyle birlikte, üretilen ürünlerin müşteriye ulaştırılması daha az bir zamanda mümkün olmaktadır. Lojistik köy öncesi
