1
T.C.
EGE ÜNİVERSİTESİ TIP FAKÜLTESİ
İÇ HASTALIKLARI ANABİLM DALI
MYELODİSPLASTİK SENDROM TANILI HASTALARDA TET2, ASXL1, IDH1/2 GEN POLİMORFİZMİ VE KLİNİKLE İLİŞKİSİNİN DEĞERLENDİRİLMESİ
DR.GÜLAY ALP
DANIŞMAN PROF. DR.GÜRAY SAYDAM
UZMANLIK TEZİ
2016 İZMİR
2
T.C. EGE ÜNİVERSİTESİ
TIP FAKÜLTESİ
İÇ HASTALIKLARI ANABİLM DALI
MYELODİSPLASTİK SENDROM HASTALARINDA TET2, ASXL1, IDH1 ve IDH2 GEN POLİMORFİZMİ VE KLİNİKLE İLİŞKİSİNİN DEĞERLENDİRİLMESİ
DR.GÜLAY ALP
DANIŞMAN PROF. DR.GÜRAY SAYDAM
UZMANLIK TEZİ
2016 İZMİR
TEŞEKKÜR
İç hastalıkları uzmanlık eğitim sürecinde bilgi ve deneyimleri ile her zaman yanımda olan ve bu mesleği sevmemi sağlayan başta Anabilim Dalı Başkanımız Prof. Dr. Fehmi AKÇİÇEK’e;
Tez çalışmam ve uzmanlık eğitimim boyunca sahip olduğu bilgi birikim ve görüşleriyle beni yönlendiren tez danışmanım Prof. Dr. Güray SAYDAM’a;
Tez sürecime desteklerinden dolayı Prof. Dr. Mahmut TÖBÜ, Prof. Dr. Murat TOMBULOĞLU, Prof. Dr. Ayhan DÖNMEZ, Doç.Dr. Fahri ŞAHİN, Uzman. Dr. Nur SOYER, Uzman. Dr.Melda CÖMERT, Uzman. Dr. Mustafa DURAN, Uzman Dr. Ayşe UYSAL ‘a;
Tezimdeki katkılarından dolayı Tıbbi Biyoloji Anabilim dalından Doç. Dr. Burçin KAYMAZ, Araş. Gör. Duygu AYGÜNEŞ’e ve tezimin tüm aşamalarında yardımlarını esirgemeyen Çağdaş AKTAN’a;
Tez jürimde yer alan ve yapıcı eleştirileri ile katkısından dolayı Prof. Dr. Mehmet Ali ÖZCAN’a
Birlikte çalışmaktan her zaman mutluluk duyduğum tüm hocalarım, doktor ve hemşire arkadaşlarım ve hastane çalışanlarına;
Projemize verdiği destek nedeniyle Ege Üniversitesi Rektörlüğü Bilimsel Araştırma Proje Fonu’na,
Eğitimim ve tez çalışmam sırasında her zaman benim yanımda olan arkadaşım Günel GULİYEVA ve canım aileme teşekkür ederim.
Gülay ALP İZMİR, 2016
ÖZET
Myelodisplastik Sendrom Hastalarında TET2, ASXL1, IDH1/ IDH2 Gen Polimorfizminin Saptanması Ve Klinikle İlişkisinin Değerlendirilmesi
Miyelodisplastik sendrom (MDS) periferik sitopeni, karakteristik morfolojik bulgular ve kemik iliğinde sitogenetik anormallikler dahil çeşitli bozuklukların olduğu heterojen bir hastalıktır. IDH1 (İzositrat dehidrogenaz; 2q33.3 lokalize), IDH2 (15q26.1), TET2 (TET ten eleven ten-onkogen aile üyesi 2; 4q24) ve ASXL1 (Additional Sex Combs-Like 1; 20q11.21). gibi bazı genlerin MDS patogenezinde rolü bildirilmiştir. MDS’de seçilen genlerin polimorfizminin ve allel sıklığının belirlenmesini amaçladık. 100 MDS hastası IDH1 rs11554137, IDH2 rs121913503, rs267606870, TET2 rs763480 ve ASXL1 rs2208131 genotipi için değerlendirildi. Periferik kandan genomik Deoksiribonükleik asit (DNA) elde edildi. Polimeraz zincir reaksiyonu (PCR) TaqMan Single-nucleotide polymorphism (SNP) genotip belirlemek için (Applied Biosystems, Foster City, CA) ABI 7500 Fast kullanılmış her mutasyonu allelik dağılımında sequence detection Systems software, SDS 2,0) dizileme sistemleri kullanılmıştır. MDS’de medyan tanı yaşı 76 olup bizim çalışmamızda medyan yaş 65 (24-89) saptandı. Tanı yaşı, cinsiyet, WHO sınıfı, Uluslararası Prognostik Skorlama Sistemi (IPSS) grubu, tanıda bakılan hemogram parametreleri [Lökosit (WBC), Hemoglobin (Hb), Trombosit (PLT)] açısından polimorfizmler (yabanıl tip, heterozigot, homozigot mutant) karşılaştırıldığında istatiksel anlamlı farklılık bulunmadı. Sadece ASXL1 heterozigot+homozigot olan grubun medyan tanı yaşı anlamlı olarak daha yüksek bulundu (p=0,048). Herhangi bir polimorfizmin prognostik etkisi saptanmadı. TET2 heterozigot polimorfik grup azasitidin (AZA) tedavi yanıtı yabanıl tipe göre istatiksel anlamlı yüksek bulundu (p=0,042) Sonuç olarak bizim çalışmamız bu genlerin polimorfizminin değerlendirilmesi için başlangıç noktası olup yeni çalışmalara rehberlik edebilir.
ABSTRACT
Analysis Of TET-2, ASXL1, IDH1-2 Gene Polymorphism And İts Correlation With Clınıc Parameters İn Patients With Myelodysplastic Syndrome
Myelodysplastic syndromes (MDS) represent heterogeneous group of disorders with a variety of features including peripheral cytopenia, characteristic morphological findings and cytogenetic abnormalities in bone marrow. Some genes are reported to be involved in the pathogenesis of MDS such as IDH1 (Isocitrate dehydrogenase; localised to 2q33.3), IDH2 (15q26.1), TET2 (TET oncogene family member 2; 4q24) and ASXL1 (Additional Sex Combs-Like 1;20q11.21). Thus, identifying the recent mutations of these genes and genotyping the patients for these selected mutations might have clinical impact in MDS. We aimed to determine the genotype distribution and allele frequency of selected genes in MDS cases. Total 100 patients were genotyped for 5 mutations as IDH1 rs11554137, IDH2 rs121913503–rs267606870, TET2 rs763480, and ASXL1 rs2208131. DNA was isolated from each patients’ peripheral blood samples. Following PCR reactions with TaqMan SNP genotyping assay (Applied Biosystems, Foster City, CA) via ABI 7500 Fast instrument, the cases were genotyped with “sequence detection Systems software, SDS 2,0) for allelic discrimination for each mutation. The median age at diagnosis of MDS is 76 in our study revealed a median age of 65 (24-89). Age at diagnosis, gender, WHO classification, IPSS group, diagnosis (WBC-Hb-PLT) compared with polymorphisms (wild-type, heterozygous, homozygous mutants) were not statistically significant differences. Only ASXL1 heterozygous group median age at diagnosis was significantly higher than homozygous (p=0,048). No significant association with the prognostic impact any of the polymorphisms. TET2 heterozygous polymorphic group of azacitidine (AZA) treatment response was significantly higher compared to the wild type. (p=0,042). Our study is the starting point for genotyping these polymorphism that might have clinical guidance for MDSs.
İÇİNDEKİLER
TEŞEKKÜR ... I ÖZET ... II ABSTRACT ... III İÇİNDEKİLER ... IV TABLOLAR LİSTESİ ... VII ŞEKİLLER LİSTESİ ... VIII GRAFİKLER LİSTESİ ... VIII KISALTMALAR LİSTESİ ... IX 1 GİRİŞ-AMAÇ ...1 2 GENEL BİLGİLER ...3 2.1 Tanım ... 3 2.2 Epidemiyoloji ... 3 2.3 Klinik Prezentasyon ... 5 2.3.1 Enfeksiyon ... 5 2.3.2 Otoimmun Anormallikler ... 5 2.4 Patolojik Özellikler ... 6
2.4.1 Tam kan sayımı ... 6
2.4.1.1 Anemi ... 6
2.4.1.2 Lökopeni ... 6
2.4.1.3 Trombositopeni ... 6
2.4.1.4 Trombositoz ... 6
2.4.2 Periferik Kan Yayması ... 7
2.4.2.1 Eritrositler ... 7
2.4.2.2 Lökositler ... 7
2.4.3 Kemik iliği aspirasyon ve biyopsisi ... 8
2.4.3.1 Kemik iliği Aspirasyonu ... 8
2.4.3.2 Kemik iliği Biyopsisi ... 8
2.6 Flow sitometri ... 9
2.7 Genetik özellikler ... 10
3 TANI VE SINIFLAMA ... 10
3.1 Sınıflama Sistemi ... 12
3.1.1 Refrakter Sitopeni Tekli Seri Displazi ... 13
3.1.2 Refrakter Anemi Ve Halka Sideroblast ... 14
3.1.3 Refrakter sitopeni çoklu seri displazi ... 14
3.1.4 Refrakter sitopeni artmış blast ... 14
3.1.5 İzole 5 q ... 14 3.1.6 Sınıflandırılamamış MDS (MDS-U) ... 16 3.2 Skorlama Sistemi ... 18 3.2.1 WPSS ... 18 3.2.2 IPSS ... 19 3.2.3 R-IPSS ... 20
3.3 MDS’de Yeni Sitogenetik Risk Sınıflaması ... 21
3.4 Tedavi ... 22
3.4.1 Eritropoez uyarıcı ilaçlar ... 22
3.4.2 İmmün supresif İlaçlar ... 22
3.4.2.1 Antitimosit Globülin-Siklosporin ... 22
3.4.2.2 Lenalidomid ... 23
3.4.2.3 Talidomid ... 23
3.4.3 Hipometile Edici İlaçlar ... 23
3.4.3.1 Azasitidin ... 23 3.4.3.2 Desitabin ... 24 3.4.4 Destek Tedavisi ... 24 3.4.5 AKİT ... 25 3.5 Genetik İnceleme ... 26 3.5.1 Metafaz Sitogenetiği ... 26 3.5.2 FISH ... 29
3.5.3 Karşılaştırmalı Genomik Hibridizasyon (CHG) ... 29
3.5.4 SNP ... 31
3.6 Mutasyonlar ... 33
3.7.1 TET2 ... 37
3.7.2 ASXL ... 41
3.7.3 IDH1 ve IDH2 ... 42
3.8 Genetik Lezyonların Tedaviye Etkisi ... 44
4 MATERYAL VE METOD ... 46
4.1 Hasta verilerinin ve Örneklerin Toplanması ... 46
4.2 Kandan DNA izolasyonu ... 46
4.3 Genler ve SNP’ ler ... 48
4.4 Gerçek zamanlı PCR Aşaması ... 48
4.4.1 PCR Reaksiyon Hacmi ... 49
4.4.2 PCR Koşulları ... 49
4.5 İstatistik ... 50
5 BULGULAR ... 51
5.1 Tanı Yaşı ve Laboratuvar Bulguları ... 51
5.2 WHO Sınıflamasına Göre Hasta Dağılımı ... 52
5.3 IPSS’ye Göre Dağılım, Lösemiye Progresyon Ve Mortalite ... 54
5.4 RIPSS’ye Göre Dağılım, Lösemiye Progresyon Ve Mortalite ... 56
5.5 Kemik İliği Selüleritesi Ve Retiküler Lif Derecesi ... 57
5.6 Klasik Sitogenetik Karyotip Sonuçları ... 57
5.7 FISH MDS paneli sonuçları ... 58
5.8 Almış Oldukları Tedaviler ... 60
5.9 Genel Sağkalım, Progresyonsuz Sağkalım Ve Mortalite ... 60
5.10 Polimorfizm Sonuçları ... 62
5.11 Hardy-Weinberg Dengesine Göre Sonuçların Değerlendirilmesi ... 62
5.12 Sonuçların Hap Map Avrupa Verileri İle Karşılaştırıması ... 63
5.13 SNP, Demografik Veriler Ve Klinikle İlişkisinin Değerlendirilmesi ... 63
5.14 SNP, Karyotip ve FISH Sonuçlarının Karşılaştırılması ... 67
5.15 Sonuçların Prognostik Etkisinin Değerlendirilmesi ... 68
5.16 Sonuçların AZA Tedavi Yanıtına Etkisi ... 71
6 SONUÇLAR ... 72
7 TARTIŞMA ... 73
TABLOLAR LİSTESİ
Tablo 1 MDS’de Sık Saptanan Sitogenetik Anormallikler; ... 10
Tablo 2 WHO 2008 MDS Tanı Kriterleri ... 12
Tablo 3 FAB ve WHO Sınıflamasının Karşılaştırılması ... 13
Tablo 4 NCCN Revizyonu Sonrası WHO (2008) MDS Sınıflaması ... 16
Tablo 5 WPSS ... 18
Tablo 6 WPSS’ye Göre Genel Sağkalım Ve Lösemiye Dönüşüm Olasılığı ... 18
Tablo 7 IPSS ... 19
Tablo 8 IPSS’ ye Göre Genel Sağkalım Ve Lösemiye Dönüşüm Olasılığı... 19
Tablo 9 R-IPSS... 20
Tablo 10 R-IPSS Göre Genel Sağkalım ve AML Dönüşüm Olasılığı ... 20
Tablo 11 MDS’de Yeni Sitogenetik Risk Sınıflaması [39] ... 21
Tablo 12 Genetik Anormallikleri Saptama Yöntemleri... 26
Tablo 14 MDS Tanımlanmış Olan Mutasyonlar ... 35
Tablo 15 Hastaların Genel Demografik Özellikleri ... 51
Tablo 16 WHO Sınıflamasına Göre Hastaların Klinik Ve Demografik Verileri ... 52
Tablo 17 WHO Sınıflamasına Göre Hastaların Karşılaştırılması ... 53
Tablo 19 FISH ve Karyotip sonuçlarının karşılaştırılması ... 59
Tablo 20 Ferritin, Albümin ve LDH Mortaliyete Etkisi ... 61
Tablo 21 Poliformizm Oranları Ve Allel Sıklığı ... 62
Tablo 22 Sonuçların Hapmap Avrupa Verileri İle Karşılaştırıması ... 63
Tablo 23 TET2 -Demografik Ve Klinik Verilerle İlişkisinin Değerlendirilmesi ... 64
Tablo 24 ASXL1- Demografik Ve Klinik Verilerle İlişkisinin Değerlendirilmesi ... 64
Tablo 25 IDH1-Demografik Ve Klinik Verilerle İlişkisinin Değerlendirilmesi ... 65
Tablo 26 IDH2-Demografik Ve Klinik Verilerle İlişkisinin Değerlendirilmesi ... 65
Tablo 27 SNP, Selülerite, Blast Sayısı, RLD, Karşılaştırılması ... 66
Tablo28 SNP, Karyotip ve FISH Karşılaştırılması ... 67
Tablo 29 TET2 Prognostik Etkisi ... 68
Tablo 30 ASXL1 Prognostik Etkisi ... 69
Tablo 31 IDH1 Prognostik Etkisi ... 69
Tablo 32 IDH2 Prognostik Etkisi ... 70
ŞEKİLLER LİSTESİ
Şekil 1 MDS’de Sitogenetik Bulguların Dağılımı [44] ... 27
Şekil 2 MDS’de DNA Metilasyonu Ve Histon Modifikasyonu [39] ... 33
Şekil 3 TET2 Geninin Kromozomdaki Yeri; 4q24 ... 37
Şekil 4 TET2 Proteinin İşlevi ... 37
Şekil 5 TET yapısı ve fonksiyonel alanları [75] ... 38
Şekil 6 TET2 rs763480 fiziksel lokalizasyonu -106,071,597 ... 40
Şekil 7 ASXL1 Geninin Kromozomdaki Yeri; ... 41
Şekil 8 ASXL1 rs 2208131 fiziksel lokalizasyonu 30,948,281 ... 42
Şekil 9 IDH1 Geninin Kromozomdaki Yeri;2q33.3 ... 42
Şekil 10 IDH2 Geninin Kromozomdaki Yeri; 15q26.1 ... 42
Şekil 11 Yabanıl Tip IDH ve Mutant IDH Aktivitesi ... 43
Şekil 12 IDH1 SNP rs11554137 fiziksel lokalizasyonu (209,113,1192) ... 44
Şekil 13 IDH2 SNP rs121913503 fiziksel lokalizasyonu (90,631,838) ... 44
Şekil 14 DNA metilasyon ve histon modifikasyon düzenleyici tedaviler [111] ... 45
GRAFİKLER LİSTESİ Grafik 1 Yaşa Göre MDS İnsidansı[13]... 4
Grafik 2 WHO Sınıflamasına Göre Hasta Dağılımı ... 52
Grafik 3 IPSS Skoruna Göre Hasta Dağılımı ... 54
Grafik 4 IPSS Skoruna Göre Lösemiye Progresyon ... 54
Grafik 5 IPSS’ye Göre Mortalite ... 55
Grafik 6 Düzenlenmiş IPSS’ye Göre Sağkalım ... 55
Grafik 7 RIPSS’ye Göre Sınıflama... 56
Grafik 8 RIPSS’ye Göre Lösemiye Progresyon ... 56
Grafik 9 RIPSS’ye Göre Mortalite ... 57
Grafik 10 Karyotip Sonuçları ... 58
Grafik 11 FİSH MDS Panel Sonuçları ... 59
Grafik 12 Genel sağkalım ... 60
Grafik 13 AMLye progresyonsuz sağkalım ... 61
Grafik 14 TET2 polimorfizm durumuna göre genel sağkalım ... 68
Grafik 15 ASXL1 Polimorfizm Durumuna Göre Genel Sağkalım ... 69
Grafik 16 IDH1 Polimorfizm Durumuna Göre Genel Sağkalım ... 70
KISALTMALAR LİSTESİ α-KG α-ketoglutarat
AKIT: Allojenik kemik iliği transplantasyonu AML: Akut miyeloid lösemi
Asx: Drosophila Additional sex combs’un ASXL: Additional sex-comb like
ATG: Anti Timosit Globülin AZA: Azasitidin
CHG: Comparative genomic hybridization CBL: Casitas B-lineage Lymphoma
MYH 11: Myosin, heavy chain 11, smooth muscle CK1A1: Kazein Kinaz 1A1
CN-AML: Sitogenetik normal AML
CPG: Metil cytosine-guanine dinucleotides Del: Delesyon
Der: Dengesiz translokasyon DFX: Deferasiroks
DFO: Deferroksamin DSBH:
DNA: Deoksiribonükleik asit
DNMTA: DNA Cytosine-5-Methyltransferase EPO: Eritropoietin
ES: Eritrosit süspansiyonu ETV6: Ets variant 6
EZH: Enhancer of Zeste homolog FAB: Fransız Amerikan İngiliz FISH: Floresan İnsitu Hibridizasyo FLT3: Fms-Related Tyrosine Kinase 3 G-CSF: Granülosit stimüle edici faktör CK1A1: Kazein kinaz 1A1
Hb: Hemoglobin
hCM: Hidroksimetil sitozin
HKHN: Hematopoetik Kök Hücre Nakli IPSS: International Prognostic Scoring System ITP: İmmün trombositopeni
IV: İntravenöz JAK2: Janus kinase 2
KLL: Kronik lenfosittik Lösemi
KMML: Kronik myelomonositer lösemi KMPH: Kronik myeloproliferatif Hastalık
KMT2A/MLL: Lysine (K)-specific methyltransferase 2A MAF: Minör Allel Frekansı
MCV: Ortalama eritrosit hacmi
MCHC: Ortalama hemoglobin konsantrasyonu mC: Metil sitozin
MDS: Miyelodisplastik Sendrom
MDS/MPN: Myelodisplastik Sendrom/Myeloproliferatif Neoplazi MDS-U: Sınıflanmamış MDS
Meis1: Meis homeobox 1
miR29B DNMT ekspresyonunu modüle eden miRNA MLL: Mixed-lineage leukemia
MNS: Mutlak Nötrofil Sayısı
NADP: Nikotinamid adenin dinükleotid
NCCN: National Comprehensive Cancer Network
NRAS: Neuroblastoma RAS viral (v-ras) oncogene homolog PCR: Polymerase chain reaction
PLCB1: Fosfoinositid-fosfolipaz C ß1 PLT: Platelet -Trombosit
PNH: Paroksismal Noktürnal Hemoglobinuri RA: Refrakter Anemi
RAEB: Artmış Blastlı Refrakter Anemi RARS: Halka Sideroblastlı Refrakter Anemi RCMD: Çoklu Dizide Displazili Refrakter Sitopeni
RB1: Retinablastom1 R-IPSS Revised IPSS RN: Refrakter Nötropeni Rpm: Revolutions per minute RPS19: RPS19 ribosomal protein S19 RT: Refrakter Trombositopeni
RT-PCR: Revesr Transkriptaz Polimeraz Zincir Reaksiyonu RUNX1:Runt-related transcription factor 1
SBDS: Shwachman-Bodian-Diamond syndrome
SPSS: Statistical Packages of Social Sciences, SPSS for Windows SRSF2 Serine/arginine-rich splicing factor 2
SRSF2: Serine/arginine-rich splicing factor 2 SF3B1: Splicing Factor 3b, Subunit 1 SNP: Single-nucleotide polymorphism TDG: DNA glikozilaz
TET: Ten eleven translocation TP53: Tumor protein p53
U2AF1:U2 Small Nuclear RNA Auxiliary Factor 1 WBC: White Blood Cells -Lökosit
WHO: Dünya Sağlık Örgütü
ZRSR2: Zinc Finger RNA-Binding Motif And Serine/Arginine Rich 2 5caC: 5-carboxylcytosine
5mC: 5-metilcytosine 5fC: 5 formylcytosine
WPSS World Health Organization classification–based Prognostic Scoring System/DSÖ sınıflamasını temel alan prognostik skorlama sistemi
1 GİRİŞ-AMAÇ
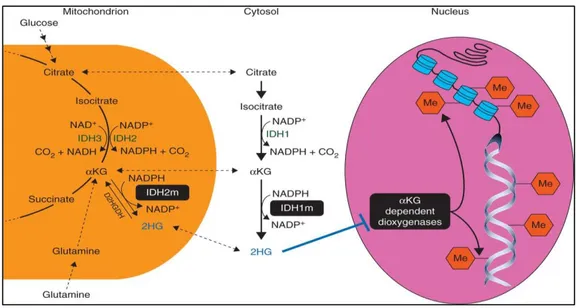
MDS etkisiz hematopoez, periferik kanda sitopeniler ve artmış Akut Myeloid Lösemi (AML) dönüşüm riski olan, klinik ve sitogenetik olarak heterojen klonal bir hastalıktır. MDS genellikle yaşlıları etkiler; olguların % 80’i 60 yaşından sonra tanı alır ve medyan tanı yaşı 76’dır [1]. Amerika Birleşik Devletleri’nde tüm yaş gruplarında sıklığı 100.000’de 3-4’tür [2]. 70 yaş üstünde sıklığı 100.000’de 22-45’e çıkmakta ve yaşla artmaktadır [2]. Ülkemizden henüz sıklığını belirleyecek geniş epidemiyolojik veri bulunmamaktadır. MDS sınıflamasında Dünya Sağlık Örgütü (WHO) 2008 sınıflaması kullanılmaktadır. WHO tekli seri displazi ile Refrakter sitopeni [ (Refakter trombositopeni (RT), Refrakter Anemi (RA), Refrakter Nötropeni (RN) ], halka sideroblast ile Refrakter anemi (RARS), çoklu seri displazi (RCMD) refrakter sitopeni blast artışı ile Refrakter anemi 1-2 (RAEB 1-2), izole del (5q) ve sınıflandırılmamış MDS (MDS-U) olarak sınıflandırır. MDS tanılı hastalarda tedavi seçiminde prognostik skorlama en önemli belirleyecidir. Bu amaçla 1997’de Uluslarası Prognostik Skorlama Sistemi (IPSS=International Prognostic Scoring System) ve 2008’de WHO Prognostik Skorlama Sistemi (WPSS-World Prognostic scoring system) oluşturulmuştur. IPSS’de karyotip (iyi-orta-kötü), sitopeniler ve kemik iliği blast oranı kullanılmaktadır. WPSS ise hastaların transfüzyon gereksinimini göz önüne alan yeni bir risk sınıflama sistemidir. IPSS daha yaygın olarak kullanılmaktadır. 2012’de yeni moleküler belirteçlerin keşfi ile oluşturulan Revize skorlama sistemi (R-IPSS-Revised İnternational Prognostic Scoring System) oluşturulmuştur ancak henüz yaygın klinik kullanıma girmemiştir [3].Risk skoruna göre hastalar gruplara ayrılmakta ve tedavileri planlanmaktadır. MDS’nin bu güne kadar patogenezi net olarak aydınlatılamamıştır. MDS hastalarının yaklaşık % 50’sinde karyotip anormalliği vardır. SNP dizi bazlı karyotipleme genomdaki küçük dengesizliklerin ve segmental uniparental dizomilerin saptamasında rol oynar. SNP’nin prognostik önemi nedeni ile konvansiyonel sitogenetiğe tamamlayıcı metod haline gelmiştir. Literatürde MDS ile ilgili çok sayıda mutasyon ve kromozomal aberasyon saptanmıştır. Mutasyona uğramış genler genellikle 4 farklı fonksiyonel gruba ait genlerdir. Bu gen grupları hücrede sitokin sinyali, DNA metilasyonu, histon modifikasyonu ve spleozom fonksiyonlarını yerine getirir. MDS’de anormal farklılaşmaya DNA metilasyon kaybı neden olmaktadır. Metilasyonda TET2 ve IDH 1-2, histon metilasyon kontrolünde ise ASLX1 rol oynamaktadır. TET2 5-metilsitozini (5mC) 5-hidroksimetilsitozine (5hmC) çevirir, alfa ketoglutarat (aKG) ve Fe (II)-bağımlı
hidroksilaz enzimlerini kodlayan bir gendir, 4q24 kromozomunda yer alır. TET2 mutasyonu MDS tanılı hastalarda ortalama % 20, Kronik myelomonositer Lösemi (KMML)’de % 30-50 oranında görülür. MDS hastalarında sık görülen TET2 mutasyonunun saptanmasının IPSS’den bağımsız olarak iyi prognoz ile ilişkili olduğunu gösteren çalışmalar mevcuttur [4]. Itzykson ve ark. (2011) yaptıkları çalışmada TET2 mutasyonu pozitif olanların mutasyon negatif olanlara göre AZA tedavisine daha iyi yanıt verdiğini göstermiştir [5]. MDS’de epigenetik anormallikler bazen erken ortaya çıkar. Örneğin TET2-ASXL1 mutasyonu AML’ye dönüşümden yıllar önce görülebilir. Fakat TET2 mutasyonu hematopoezi bozulmuş ancak aşikar MDS’si olmayan yaşlı bireylerde de bildirilmiştir [5]. ASXL1 histon modifikatörleri ile etkileşir ve 20q11.21 kromozomunda yer alır. ASXL1 hastaların % 10-20’sinde KMML’de % 40 oranında saptanmıştır[6]. ASXL1, TET2 den sonra en iyi tanımlanmış ve en sık görülen mutasyon olup, düşük riskli hastalarda kötü prognostik göstergedir. Thol ve ark. (2011) AML’ye dönüşüm süresinin bu hastalarda daha kısa olduğu göstermiştir. IDH izositratı α-KG’a çeviren Nikotinamid adenin dinükleotid (NADP+) bağımlı krebs siklusu enzimidir. IDH-1 2q33.3, IDH2 15q26.1 kromozomunda yer alır. TET2 için gerekli α-ketoglutaratı (α-KG) üretir ve mutasyonu hastaların < % 10’unda (% 4-12) saptanır. Mutasyona uğramış IDH1-2 proteinlerinin ürettiği 2 hidroksiglutarat’ın (2-HG) TET2 gibi α-KG bağımlı enzimleri inhibe etttiği ve bununda DNA hipermetilasyonu ile sonuçlandığını gösterilmiştir [7]. IDH mutasyonunun prognozdaki etkisini değerlendirme amaçlı yapılan bir meta-analizde ise orta-1 MDS grubunda total sağkalımı kötü etkilediği gösterilmiştir [7]. Yeni epigenetik belirteçler MDS için kullanılan sınıflama sistemine dahil edilip prognozu belirlemede faydalı olabilir. Ayrıca ileri çalışmalar ile epigenetik regülatörleri etkileyen tedaviler geliştirilebilir. Klinik kullanımda olan ve etkinliği gösterilmiş ilaçlardan AZA ve Desitabin DNA metilasyon inhibitörleridir [8].Histon modifikatörleri Histon deasetilaz (HDAC) inhibitörleri üzerinden etkili ilaçların çalışmaları devam etmektedir. MDS’de metilasyon genlerinin somatik mutasyonu ile ilgili çok sayıda çalışma olmasına rağmen bu genlerin IDH1 hariç polimorfizmini değerlendiren ülkemizde ve dünyada çalışma yoktur. Biz çalışmamızda MDS tanısı ile takip edilen ya da yeni tanı konulan hastaların periferik kanlarından DNA izole ederek; TET2, ASXL1, IDH1-2 gen polimorfizm durumunu saptamayı ve klinikle ilişkisini değerlendirmeyi amaçladık.
2 GENEL BİLGİLER
2.1 Tanım
MDS periferik kanda sitopeniler (kemik iliğinde hipersüleriteye rağmen) ve bir veya daha çok seride displazi ile karakterize değişken akut lösemiye transformasyon riski olan klonal hematopoetik kök hücre hastalığıdır. Trombosit, granülosit ve eritrositlerin üretiminde değişken bir azalma vardır. Hematopoez kantitatif olarak artmasına rağmen periferik kanda sitopenilerin olmasının nedeni niteliksel olarak bozuk olmasıdır. MDS de novo yada bir mutajenik tedaviye sekonder (radyoterapi, kemoterapi) ortaya çıkabilir. Bu niteliksel ve niceliksel anormallikler genellikle anemi, kanama ve artmış enfeksiyon riski ile sonuçlanır.
2.2 Epidemiyoloji
De novo MDS insidansı kesin değildir ancak kanser veri tabanlarına göre konservatif tahminler Amerika birleşik devletlerinde her yıl 10.000 yeni vaka olduğunu düşündürmektedir [1]. Bir seride 100.000 de 4,1 insidans oranı bildirilmiştir [9]. Buna karşılık doğu avrupada 100.000 de 0,27 insidans oranı bildirilmiştir, belki bu farklılık hastanelerin kullandığı tanı kriterleri ve kayıt sistemi ile ilgili olabilir [10].MDS’ nin gerçek sıklığı muhtemelen daha yüksektir çünkü şüpheli vakalara komorbiditeleri nedeni ile kesin tanı testi (kemik iliği biyopsisi gibi) yapılamayabilir veya nonspesifik semptomlar hastalığın erken aşamasında gözden kaçmasına neden olabilir [11]. MDS genellikle yaşlıları etkiler; olguların % 80’i 60 yaşından sonra tanı alır ve medyan tanı yaşı 76’dır. Erkeklerde kadınlardan (yılda 100.000 kişide E/K; 4,5/ 2,7) daha yüksek insidans oranı saptanmıştır. [1]. Tedaviye sekonder MDS hariç, 50 yaşın altında görülmesi olağandışıdır [12]. MDS gelişme riski yaşla beraber artar. Williamson ve ark. (1994) 100.000 de yıllık insidansı bireylerin yaşı <50 altında; 50 -59; 60-69; 70-79;ve >80 ayrıldığında sırasıyla 0.5, 5.3, 15, 49, ve 89 olarak tahmin edilmiştir [13].
Grafik 1 Yaşa Göre MDS İnsidansı[13]
MDS çevresel faktörlerle ilişkilendirilmiştir; bunlar kimyasal maruziyeti (özellikle benzen maruziyeti), radyasyon, sigara ve kemoterapi ilaçlarına maruziyet kalıtsal genetik anomaliler (Bloom sendromu, Fanconi sendromu, Ataksi telenjektazi, Trizomi 21) ve diğer bening hematolojik hastalıklardır (Konjenital nötropeni). Raynoud fenomeni, Sjögren sendromu, inflamatuar barsak hastalığı, Behçet ve glomerulonefrit ile birlikteliği raporlanmış olmasına rağmen nedensel bir ilişki kurulamamıştır [14].
0
0
2
1
2
4
4
9
16
26
52
59 61
34
10
1
0
10
20
30
40
50
60
70
20- 25- 30- 35- 40- 45- 50- 55- 60- 65- 70- 75- 80- 85- 90-
95-Ynei ge
li
şe
n
vak
a
Yaş aralığı
2.3 Klinik Prezentasyon
MDS’nin spesifik belirti ve semptomu yoktur. Çoğu hasta tanıda asemptomatik ve rutin kan sayımında bulunan anormallikle tanı alır. Ya da sitopenilere bağlı komplikasyonlarla tanı alır (kanama, enfekiyon gibi). Anemi en sık sitopenidir ve halsizlik, anjina, baş dönmesi, egzersiz intoleransı ve kognitif fonksiyon bozukluğu ile prezente olur. Ateş ve kilo kaybı gibi sistemik semptomlar yaygın değildir genel olarak hastalığın geç bulguları ve buna bağlı komplikasyonları temsil eder. Fizik muayene bulguları nonspesifiktir, % 60 solukluk ve % 26 oranında peteşi-purpura görülmektedir [15]. Hepatomegali, splenomegali ve lenfadenopati nadirdir. Sweet sendromu başlangıç semptomu olabilir. 2.3.1 Enfeksiyon
MDS’de enfeksiyon nötropeni ve bozulmuş granülosit fonksiyonu ile ilişkilidir. En yaygın cilt olmak üzere bakteriyel enfeksiyonlar daha sıktır. Viral, fungal ve mikobakteriyel enfeksiyonlar da görülmesine rağmen eşlik eden immünsupresif tedavi yokluğunda nadirdir. Monosit spesifik esteraz artmışken myeloid hücrelerde myeloperoksidaz ve alkalen fosfotaz aktivitesi azalmış olabilir. Bunun sonucu olarak granülosit fonksiyonu, fagositoz, adezyon, bakterisidal aktivite ve kemotaksis bozulur, bakteriyel enfeksiyonlara karşı direnç azalır. Olguların çoğunda lenfositler malign klondan kaynaklanmasada MDS hastalarında da kazanılmış immün sistem bozuklukları bulunabilir. Lenfopeni CD4 lenfositlerin azalması nedeniyle transfüzyon sayısı ile ters orantılıdır. Bununla birlikte CD8 sayıları normal ya da hafifçe artmıştır [16]. İmmünglobülin üretimi değişken derecede etkilenir; hipogamaglobülinemi, poliklonal hipergamaglobülinemi ve monoklonal gamapati sırasıyla hastaların % 13, 30 ve 12’sinde görülür [17].
2.3.2 Otoimmun Anormallikler
Otoimmun anormallikler MDS’de nadir olmasına rağmen hastalığın gidişatını kötüleştirir. En sık % 7 romatizmal kalp kapak hastalığı, % 6 romatoid artrit, % 6 pernisiyöz anemi, %4 psöriyazis ve polimyaljika romatika görülmektedir. Diğer otoimmun anormallikler; sweet sendromu, perkardit, plevral efüzyon, cilt ülserleri, iritis, myozit, periferik nöropati ve kırmızı hücre aplazisidir. Hastalar bazen kutanöz vaskülit, ateş, artrit, periferal ödem ve pulmoner infiltrat ile karakterize bir klinik sendrom olarak ortaya çıkabilir [14].
2.4 Patolojik Özellikler
MDS anormal hücre morfolojisi ve bir veya birden fazla periferik kan-kemik iliği elemanlarında sayısal değişiklikle karakterize bir hastalıktır.
2.4.1 Tam kan sayımı
Tam kan sayımında makrositik veya normositik anemi, lökopeni- trombositopeni izlenebilir. Tanı anında vakaların yarısında pansitopeni mevcuttur. İzole anemi nadir olmamakla birlikte hastaların %5’den azında izole nötropeni, trombositopeni ve aneminin eşlik etmediği monositoz görülür.
2.4.1.1 Anemi
Anemi nerdeyse hepsinde uygun olmayan düşük retikülosit cevabı ile ilişkilidir. Ortalama eritrosit hacmi (MCV) makrositik (>100 fentolitre) veya normaldir. Eritrosit dağılım genişliği eritrosit hücre boyutunda artmış değişkenliği gösterir ve sıklıkla artmıştır. Ortalama hemoglobin konsantrasyonu (MCHC) eritrosit hemoglobinin hücre boyutuna oranını yansıtır ve genellikle normaldir.
2.4.1.2 Lökopeni
Hastaların yaklaşık yarısında genellikle mutlak nötropeniden kaynaklanan lökosit sayısı azalmıştır [18]. Dolaşımda immatüre nötrofiller( myelosit, promyelosit ve myeloblast) olabilir ancak blast sayısı lökosit sayısının % 20’ sinden az olmalıdır.
2.4.1.3 Trombositopeni
Değişen derecelerde trombositopeni kabaca hastaların % 25’inde vardır [18]. Aneminin aksine izole trombositopeni MDS’nin yaygın bir erken belirtisi değildir [19]. Ancak delesyon 20 q’ nun izole karyotipik anormallik olduğu hastalarda çok az morfolojik displazi ile beraber trombositopenik prezentasyon bildirilmiştir. Böyle hastalar kolaylıkla İmmün trombositopeni (ITP) ile karıştırılırlar.
2.4.1.4 Trombositoz
MDS’de trombositoz trombositopeniden daha az sıklıkla görülür. Trombositoz izole 5q sendromunda 3q21q26 ile ve refrakter ring sideroblast ve trombositoz (RARS-T) Janus kinase 2 (JAK2) mutasyon aktivitesi ile ilişkilidir.
2.4.2 Periferik Kan Yayması
Periferik kan yayması genellikle kırmızı ve beyaz hücre serisinde displazi bulguları gösterir. Trombositler morfolojik olarak genellikle normaldir. Daha az sıklıkla normalden küçük veya büyük olabilir ya da granülleri fazla olabilir, megakaryosit fragmanları görülmeyebilir.
2.4.2.1 Eritrositler
Eritrositler refrakter anemi ring sideroblastta hipokrom mikrositer olmasına karşın veya genellikle normositik ve makrositiktir [20]. Ovalomakrositoz eritrositlerde en iyi tanımlanmış morfolojik anormalliktir. Bazı vakalarda gözyaşı hücreleri, stomasitler, akantositler, eliptositler gibi hücre içi iskelet anormalliklerini yansıtan değişiklikler baskın olabilir [21]. Periferik yaymada bazofilik noktalanma, Howell–Jolly cisimcikleri ve megaloblastik çekirdekli eritrositler bulunabilir. Bu periferik kan bulguları, megaloblastik özellikler ile beraber eritroid hiperplazi, kemik iliği öncüllerinin diseritropoetik özellikleri; nükleer tomurcuklanma, karyoreksis, multinükleasyon ve sitoplazmik vakualizasyon ile karakterize gecikmiş ve bozulmuş nükleer olgunlaşma ile ilişkilidir [20]. Retikülositoz üzerine eklenen bir otoimmun hemolitik aneminin sonucu veya psödoretikülositoz olarak adlandırılan gecikmiş bir retikülosit matürasyon göstergesi olabilir.
2.4.2.2 Lökositler
Periferik kan yaymasında sıklıkla displastik nötrofiller bulunur. Bu hücreler anormal nükleer lobülasyon ve granülerite ve artan boyut gösterebilirler. Genellikle azalmış ya da hiç olmayan granülasyonun eşlik ettiği granülositlerin azalmış segmentasyonu Psödo-Pergel-Huet olarak adlandırılır. Granülositlerin topaklaşan bir kromatin paterni vardır, burada kromatin nükleer materyaldeki bir boşlukla ayrılır ve nükleer bölünme ve segmentsiz bir görünüm yaratır. Halka şeklindeki çekirdekler özellikle tedavi ilişkili MDS’de tespit edilebilir. Myeloblastlar artmış çekirdek/sitoplazma oranı, kolayca şeçilen nükleolus, ince nükleer kromatin, sitoplazmik bazofili, sitoplazmik granüllerin azalması ya da hiç olmaması gibi nükleer ve sitoplazmik özellikleri göre tespit edilir [22]. Nadiren Aurer body görülebilir. MDS ön tanısı olan hastada Aurer rod varlığı AML’ye dönüşüm habercisidir.
2.4.3 Kemik iliği aspirasyon ve biyopsisi 2.4.3.1 Kemik iliği Aspirasyonu
Optimal kemik iliği aspirasyonu blast ve diğer hücrelerinin ayrıntılı histolojik değerlendirmesi ve blast yüzdesi vermek için en az 500 hücre içermelidir. Bozulmuş myeloid olgunlaşma genellikle kolaylıkla anlaşılır. Granülosit öncülerinin yüzdesi artmış olabilir ve myeloid seride göreceli bir olgunlaşma duraksaması görülür. Sitoplazma olgunlaşması ve nükleusa göre daha hızlı ilerleyebilir [23]. Myeloid eritroid oranı değişkendir, genellikle azalmıştır. Olgunlaşmamış öncüllere doğru kayma olur ancak blast yüzdesi tanımı gereği %20 den azdır [23]. Eritrositlerde morfolojik anormallikler artmış hücre boyutu, nükleer multilokülasyon ve nükleer tomurcuklanmayı içerir. Eritroid öncülerin sitoplazmaları kaba ve kalın Periyodik asit-Schiff-pozitif granüller veya çekirdeği çevreleyen demir yüklü mitekondri gibi vakuolizasyon gösterebilir. Granülosit öncüleri de anormal hücre büyüklüğü, nükleer şekil ve artmış ya da azalmış sitoplazmik granülerite gibi displastik değişiklikler gösterebilir.
2.4.3.2 Kemik iliği Biyopsisi
Kemik iliği biyopsisi süreç ile ilgili histolojik bilgi, genel bir fikir verir. Biyopside blast yüzdesi vermek için en az 200 farklı lökosit değerlendirilmelidir. Selülerite genellikle artmıştır, normal ya da azalmış olabilir. Reaktif lenfositoz, mastositoz, lenfoid agregatlar, artmış histiyositler gibi başka özellikler de içerir. Ek olarak immatür hücreler kemik iliğinde merkez yerine endotel yüzeyi boyunca bulunur. Kemik iliği tekli ya da çoklu seride displazi özelliği gösterir.[24]. Periferik pansitopeni paradoksu; hipersülüler kemik iliği varlığına rağmen intramedüller hücre ölümü ile prematür hücre kaybını yansıtır [25]. Hiposelülerite nadir olmasına rağmen en sık olarak tedavi ilişkili MDS’de görülmektedir, aplastik anemiden ayırt edilmelidir.
2.4.3.2.1 Eritrositler
Ring sideroblastlar demir tespiti için boyanmış kemik iliği örneklerinde demir yüklü mitekondri içerir. İnternükleer köprüleme kromatin iplikçikleri ile bağlı ayrılamamış çekirdek ile karakterizedir [26]. Eritroid hiperplazi inefektif eritropoez ile bağlantılı baskın bulgu olmasına rağmen nadiren hipoplazi ya da aplazisi görülebilir.
2.4.3.2.2 Megakaryositler
Megakoryositler normal ya da sayıca artmış olabilir bazen küme meydana getirirler. Anormal megakaryositler mikromegakaryositler, geniş mononükleer formlar, birden fazla dağınık çekirdekleri olan megakaryositler ve hipogranüler megakaryositler sıklıkla kemik iliğinde bulunabilir. Mononükleer ve non lobüle formlar 5q sendromu ile birlikte saptanabilir.
2.4.3.2.3 Fibrozis
Hafifden orta dereceye kadar fibrozis MDS hastalarının %50’sinde bildirilmiştir, belirgin fibrozis ise % 10-15 bulunur [27]. Fibrozis MDS’ nin tüm alt tiplerinde görülür iken tedavi ilişkili MDS’ de daha yaygındır. Önemli bir şekilde trikrom boyası ile tespit edilen kollajen birikimi MDS’de nadirdir. Bunun yerine fibrozis derecesi artan ve kalınlaşan retikülin liferden kaynaklanır en iyi gümüş boyası ile boyanır [28]. Fibrozis çok belirgin ise MDS- Myeloproliferatif Neoplazi (MPN) çakışma sendromu olarak adlandırılır.
2.5 Histokimya ve İmmünhistokimya
İmmunhistokimyasal çalışmalar myeloid maturasyon antijenlerinde azalma ya da kayıp gösterir. Myeloperoksidaz ve alkalen fosfotaz aktivitesi myeloid hürelerde azalmış iken monositik esteraz artmış olabilir. Demir boyası ring sideroblastları tanımlar, PAS boyası eritrositlerde diseritropoez değerlendirmesi için, Peroksidaz ve Sudan black ise myeloid blastları doğrulamak için kullanılır. Nonspesifik esteraz granülosit ve monosit formlarını ayırt etmek için kullanılır. Myeloid öncüllleri ve blastları ölçmek için CD34, CD117, CD13, CD14 ve CD33 antikorları kullanılır. Displastik ve olgunlaşmamış megakaryositler von Willebrand factor, factor VIII, CD41 ve CD61 seçicilik gösteren antikorlar ile saptanır [29].
2.6 Flow sitometri
Periferik kan ve kemik iliğinin morfolojik analizinde displazi kanıtları MDS tanısında en önemli faktördür [30]. Çoklu parametreli flow sitometri sistemleri displaziyi skorlamak için geliştirilmiştir [31]. Flow sitometri bulguları MDS varlığı ve klonaliteyi gösterebilir. Bu bulgular tanısal kabul edilemezken, şüpheli durumlarda tanıyı destekler.
2.7 Genetik özellikler
Bazı kromozomal anormalliklerin rutin sitogenetik analiz esnasında tespitinde kullanılan revers transkriptaz zincir reaksiyonu (RT-PCR) ya da floresan insitu hibridizasyon (FISH) MDS sınıflamasında ve MDS-AML ayrımında yardımcıdır, prognostik risk grubu ve tedavinin belirlenmesinde en önemli faktördür [32]. Blast oranından bağımsız t(8;21)(q22;q22), inv(16)(p13.1q22) ya da t(16;16)(p13.1;q22) ve t(15;17)(q22;q21.1) saptanırsa AML tanısı koyulur. Açıklanamayan refrakter sitopeni ve morfolojik displazi yokluğunda aşağıdaki kromozomal anormalliklerden birinin varlığı MDS kanıtıdır [33].
Tablo 1 MDS’de Sık Saptanan Sitogenetik Anormallikler;
●-7/del(7q) ● del(17p) / t(17p) (dengesiz translokasyon)
veya i(17q) (17p delesyonu )
●-5/del(5q) ●t(11;16)(q23;p13.3) ●del(13q) ●t(3;21)(q26.2;q22.1) ●del(11q) ●t(1;3)(p36.3;q21.3) ●del(12p) yada t(12p) ●t(2;11)(p21;q23.3) ●del(9q) ●inv(3)(q213q26.2) idic(X)(q13 ●t(6;9)(p23.3;q34.1)
Kromozomal anormallikleri tespit etmek için FISH, karşılaştırmalı genomik hibridizasyon (CGH/ Comparative Genomic Hybridization), ve heterozigotluk kaybında SNP kullanılabilir. Bazı merkezler MDS tanısında yeni nesil sekanslama kullanmaktadır, önümüzdeki yıllarda bilinen veya şüphe edilen MDS hastalarında standart çalışmaların bir parçası olacağı düşünülmektedir [34].
3 TANI VE SINIFLAMA
MDS tanısı açıklanamayan sitopenisi ve monositozu olan her hastada düşünülmelidir. İyi bir öykü ile beslenme durumu, alkol veya uyuşturucu kullanımı, ilaçlar, toksik
kimyasallara mesleki maruziyet, önceki antineoplastik ajan ya da radyoterapi görmüş olmak ve HIV enfeksiyonu veya HIV tedavisi açısından değerlendirilmelidir. Periferik yayma kemik iliği aspirasyon ve biyopsisi MDS tanısı için anahtar rol oynar. MDS ile ortak bulguları olan B12, folat ve bakır eksikliği ile çinko fazlalığı gibi durumlar dışlanmalıdır. Periferik kan yayması ve kemik iliği dikkatlice tek ya da tüm serilerde displazi bulgularını değerlendirmek için incelenmelidir. Biyopsi; süreç ile ilgili spesifik histolojik özelliklerin (fibrozis gibi) tutulum derecesine genel bir bakış sağlar. MDS tanısı için üç kriter aranır hepsi olmak zorunda değildir. Birincisi açıklanamayan sitopenilerdir. Sitopenileri tanımlamak için kullanılan değerler; Hemoglobin <10 gr / dl mutlak nötrofil sayısı <1.8 x 109 / L trombositler <100 x 109/L. Ancak sitopenilerin bu eşikleri karşılamaması eğer morfolojik displazinin kesin kanıtları varsa MDS tanısını dışlamaz. Daha önce bahsedilen spesifik genetik anormalliklerin birinin olmasıda açıklanamayan sitopenilerde MDS tanısını koydurabilir.
Tablo 2 WHO 2008 MDS Tanı Kriterleri
A- Ön kriterler
≥ 6 ay süreli sitopeniler
Nötropeni (< 1500/mm3), anemi (Hb <11g/dL), trombositopeni ( < 100.000/mm3) Sitopeni nedeni olabilecek klonal/non klonal hastalıkların dışlanması
B- Kesin kriterler
Morfolojik displazi (≥ 10%)
Myeloid, eritroid veya megakaryositer dizilerin en az birinde ≥ %10 displastik hücre veya %15 “ring” sideroblast (ifade açık değil)
Blast oranı (%1-19)
Tipik sitogenetik anomaliler (+8,-7, 5q-, 20 q-, diğer) MDS tanısı için “en az 1 adet B + 2 adet A kriteri” gereklidir
3.1 Sınıflama Sistemi
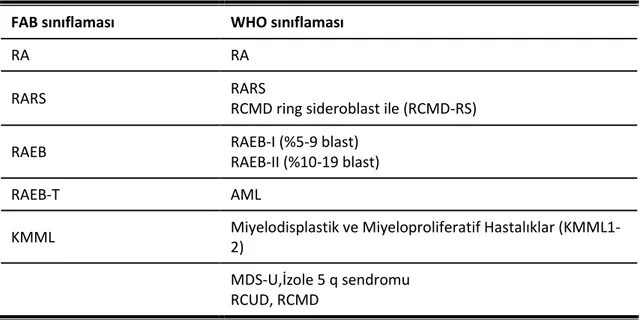
Fransız Amerikan İngiliz (FAB) sınıflama sistemi MDS için kullanılan ilk şemalardandır [35]. Kategoriler monosit ve blast sayısı halka sideroblast olup olmaması gibi sadece morfolojik özelliklere dayanmaktadır. 1982 yayınlanmış olup FAB sınıflamasında kategoriler RA, RARS, RAEB, Transformasyonda Blast Artışlı Refrakter Anemi (RAEB-T), KMML olarak tanımlanmıştır. Bu sınıflamada, akut lösemi tanısı için kemik iliği ya da periferik kanda %30’dan fazla miyeloblast bulunması gerekmektedir. %30 veya daha az miyeloblast bulunan hastalar miyelodisplazi olarak sınıflandırılmıştır. İlk iki kategori olan RA ve RARS, kemik iliğinde % 5’den az miyeloblast bulunmasıyla karakterizedir. RA ve RARS’lı hastalarda ortalama sağkalım daha uzundur ve diğer kategorilere göre akut lösemiye ilerleme daha düşük orandadır. RAEB ve RAEB-T’li hastaların kemik iliğinde sırasıyla % 5-20 ile % 21-30 miyeloblast bulunmaktadır. Bu sınıflamanın kısıtlılıkları RAEB-T ve AML benzer biyolojik süreçleri göstermektedir. Ek olarak RA ve RARS kategorilerinde displazi varlığı değerlendirilmemiştir. Ayrıca sitogenetik özellikleri (5q gibi) içermemektedir. 2008’de WHO sınıflaması geliştirilmiştir En önemli farklılıkları; KMML’nin MDS alt tiplerinden çıkarılıp myeloproliferatif hastalıklara eklenmesi, RAEB-T’nin AML olarak kabul edilmesi ve sitogenetik olarak izole 5 q’nun ayrı bir alt grup olarak tanımlanmasıdır. 2009’da revize edilerek tekli seri ve çoklu seri displazi alt grupları eklenmiştir.
Bu sınıflama tamamen histolojik ve morfolojik temelli değildir daha doğru tanı ve sınıflama sistemi için sitogenetik bulgular entegre edilmelidir. MDS’de yaygın genetik değişiklikler sitogenetik, epigenetik ve RNA’da submikroskopik olarak farklı test metodları gerektiren düzeylerde olur. Bu testler PCR metodlarına dayanmaktadır.
Tablo 3 FAB ve WHO Sınıflamasının Karşılaştırılması FAB sınıflaması WHO sınıflaması
RA RA
RARS RARS
RCMD ring sideroblast ile (RCMD-RS)
RAEB RAEB-I (%5-9 blast)
RAEB-II (%10-19 blast)
RAEB-T AML
KMML Miyelodisplastik ve Miyeloproliferatif Hastalıklar (KMML1-2)
MDS-U,İzole 5 q sendromu RCUD, RCMD
MDS/MPN sınıflamasında KMML1-2, Atipik KML, Jüvenil Miyelomonositik Lösemi, RARS-T ve MDS/MPN çakışma sendromu yer almaktadır. Yaygın kullanımı nedeni ile bundan sonra hastalar WHO sınıflamasına göre değerlendirilecektir. WHO sınıflaması ise morfoloji, immünfenotipleme, sitogenetik ve klinik özelliklere göre yapılmıştır. Karışıktır ve uzman bir hematopatoloğun morfolojik değerlendirmesi gerekir. WHO sınıflama sistemi tahmini aşağıdaki yüzdeler ile 6 gruba ayırır. RCUD < %5, RARS < %5, RCMD %70, RAEB % 25, MDS izole 5 q % 5 ve MDS-U <% 5 oranında izlenir.
3.1.1 Refrakter Sitopeni Tekli Seri Displazi
Kemik iliğinde %5’den az periferik kanda % 1’den az blastla karakterizedir. Monositoz, önemli sayıda halka sideroblast ve Aurer body yoktur. Displaziyi tanımlamak için önerilen seviye %10’un üzerinde olmasıdır. Refrakter anemi için hb<10 g/dL, refrakter trombositopeni için trombosit <100.000 /microL ve refrakter nötropeni için nötrofil<1800 /microL olmalıdır. Morfolojik displazi ve sitogenetik anormallikler varsa bu değerlerin üzerindeki seviyeler olması MDS’yi dışlamaz. Hastaların çoğu tek seride displazi ve sitopeni gösterirken, iki seride sitopeni ve tek seride displazi olanlar da bu gruba dahil edilir.
Refrakter pansitopenisi ve tek seride displazisi olan hastalar RCUD olarak kabul edilmemeli MDS sınıflandırılamamış kategorisine alınmalıdır.
3.1.2 Refrakter Anemi Ve Halka Sideroblast
RARS, RA kriterlerini karşılayan tüm hastalarda aynı zamanda kemik iliğinde >%15 oranında halka sideroblast görülmesidir. Patolojik sideroblastlar hücre başına >5 demir yüklü mitekondri içerir ve demir tespiti için boyanmış kemik iliği örneklerinde belirgin olabilir. Beşten fazla demir yüklü mitekondri içeren sideroblastlar nükleer kenarın üçte birinden fazlasını işgal ederse halka olarak adlandırılır. Ring sideroblast veya demir yükü MDS alt tiplerinden herhangi birinde bulunabilir ancak RARS’ın öncelikli özelliğidir. RARS genellikle iyi prognozludur.
3.1.3 Refrakter sitopeni çoklu seri displazi
Kemik iliğinde % 5’den az blast ve ≥ 2 miyeloid dizide (nötrofil ve/veya eritroid öncül ve/veya megakaryosit) ≥ % 10 displazi ile karakterizedir. % 15 halka (ring) sideroblast eşlik ederse RCMD-RS olarak adlandırılır. Periferik kanda sitopeniler olabilir, blast sayısı <%1 olmalıdır.
3.1.4 Refrakter sitopeni artmış blast
Sitopeniler, tek veya çoklu seride displazi ile beraber RAEB kemik iliğinde >%5 blast oranı ile karakterizedir. %5-9 arasında blast oranı RAEB-1, 10-19 arasında blast oranı ise RAEB-2 olarak ikiye ayrılır. RAEB-1 ve RAEB-2 olanlar arasında blast sayısından başka anlamlı fark yoktur ancak RAEB-2 daha kısa medyan sağkalım ve daha fazla artmış AML riski taşımaktadır. RAEB-2’ de Aurer body kemik iliği veya periferik kanda görülebilir.
3.1.5 İzole 5 q
MDS tanılı hastaların yaklaşık %5’ini şiddetli anemi, korunmuş trombosit sayısı ve tek sitogenetik anormallik olan 5. kromozomun uzun kolunun intertisyel delesyonu ile karakterize 5q sendromu oluşturur [36]. 5 q sendromu nispeten iyi huylu bir seyir izler. Akut lösemiye dönüşüm insidansı düşüktür ve lenalidomid gibi yeni ajanlarla tedaviye duyarlıdır. İzole 5 q’nun lenalidomid yanıtı 5.kromozomun yaygın olarak silinmiş bölgesinde bulunan kazein kinaz 1A1 (CK1A1) geninin tek kopyasının silinmesi ile açıklanabilir [37]. CK1A1 olan hücreler lenalidomid tarafından öldürülmeye alışılmadık şekilde duyarlıdır. 5 q primer MDS’
den farklı olarak kadınlarda daha sık, medyan tanı yaşı 65-70 arasındadır [33]. Etkilenen hastalar genellikle dirençli makrositik anemi, normal ya da yüksek trombosit sayısı ve anlamlı nötropeni olmaması ile karakterizedir [38]. Kemik iliği tek loblu ya da iki loblu çekirdekleri olan mikromegakaryositler ile karakterizedir. Hastaların % 80’ninde kemik iliğinde %5 den az blast vardır [38].
3.1.6 Sınıflandırılamamış MDS (MDS-U)
Kronik sitopeniler periferik kanda %1’den az blast kemik iliğinde % 5’den az blast olması MDS sınıflandırılamayan tip olarak kategorize edilir.
Tablo 4 NCCN Revizyonu Sonrası WHO (2008) MDS Sınıflaması
Alttip Periferik Kan Kemik İliği
Tek dizide displazinin olduğu refrakter sitopeni (Refractory cytopenia with unilineage dysplasia = RCUD)
- Refrakter anemi (RA) - Refrakter nötropeni (RN) -Refrakter trombositopeni (RT)
Tek veya iki dizide sitopeni*
Blast < % 1†
Tek dizide displazi; Etkilenen dizide ≥ % 10 displazi
Blast < % 5
Halka (ring) sideroblast < % 15
Halka (ring) sideroblastlı refrakter anemi (RARS)
Anemi var Blast yok
Sadece eritroid displazi Halka (ring) sideroblast ≥ % 15
Çoklu dizide displazili refrakter sitopeni (Refractory cytopenia with multilineage dysplasia = RCMD)
Sitopeni (ler) Blast < % 1† Auer cisimciği yok Monosit< 1000/mm3
≥ 2 miyeloid dizide (nötrofil ve/veya eritroid
öncül ve/veya
megakaryosit) ≥ % 10 displazi Blast < % 5 Auer cisimciği yok ± % 15 halka (ring) sideroblast
Artmış blast sayılı refrakter anemi-I (Refractory anemia with excess blast-I = RAEB-I)
Sitopeni (ler) Blast ≤ % 2-4† Monosit< 1000/mm3
Tek veya çok dizide displazi
% 5-9 blast† Auer cisimciği yok Artmış blast sayılı refrakter anemi-II
(Refractory anemia with excess blast-II = RAEB-II)
Sitopeni (ler)
Monosit<1000/mm3 Blast<% 5-19‡ ± Auer cisimciği‡
Tek veya çok dizide displazi % 10-19 blast‡ ± Auer cisimciği‡ Sınıflandırılamamış MDS (MDS unclassified = MDS-U) Sitopeniler
Tek dizide displazi var veya displazi olmaksızın MDS sitogenetiği var Blast < % 5
MDS ile ilişkili izole 5q delesyonu
Anemi Normal/artmış trombosit Eritroid displazi Blast < % 5 İzole 5q delesyonu Transformasyon gösteren artmış blast
sayılı refrakter anemi (Refractory anemia with excess blast in transformation = RAEB-T)
Sitopeniler, %5-19 blast
Çoklu dizide displazi, Auer cisimciği ±, Blast %20-30
NCCN: National Comprehensive Cancer Network*Bisitopeni gözlenebilir ancak pansitopeni varsa MDS-U olarak sınıflandırılmalıdır.
† Kemik iliğinde myeloblast yüzdesi>5 ancak periferik kanda %2-4 blast varsa RAEB-1 kabul edilmeli , % 1 Myeloblast olan RCUD-RCMD ise MDS-U kabul edilmelidir.
‡ Aurer rod ile birlikte kanda <%5 kemik iliğinde >%10 blast olanlar RAEB-2 kabul edilir. Periferik kanda %5-19 blast olan RAEB-2 kabul edilirken Aurer rod ve /veya kemik iliğinde %10-19 blast varsa periferik kanda blast <%5 olabilir. Benzer şekilde RAEB-2 vakalarında kemik iliğinde<%10 blast ancak periferik kanda Aurer rod ve/ veya %5-19 blast olabilir. .
3.2 Skorlama Sistemi
Yeni tanı almış MDS hastalarının prognozunu belirlemek için skorlama sistemleri kullanılmaktadır. IPSS MDS hastalarında en yaygın kullanılan skorlama sistemidir. İlk olarak 1997 yılında yayınlanmıştır. WPSS sınıflama sistemide mevcuttur ancak yaygın kullanılmamaktadır. R-IPSS ise yeni tanımlanan skorlama sistemidir.
3.2.1 WPSS
WHO sınıflamasına, sitogenetik ve transfüzyon ihtiyacına göre WPSS sınıflaması sistemi oluşturulmuştur ancak yaygın klinik kullanıma girmemiştir.
Tablo 5 WPSS
Prognostik Kategori WPSS Prognostik Skor Değeri
0 1 2 3
WHO kategorisi RCUD, RARS
izole 5q
RCMD RAEB-1 RAEB-2
Sitogenetik1 İyi Orta Kötü (-)
Ağır Anemi* Yok Var (-) (-)
1
Sitogenetik;
İyi: Normal, -Y, izole del (5q), izole del (20q);
Kötü: Kompleks (≥3 anormallik) veya kromozom 7 anormalliği; Orta: diğer anormallikler
*Kadında Hb<8, erkekte <9 g/dL
Tablo 6 WPSS’ye Göre Genel Sağkalım Ve Lösemiye Dönüşüm Olasılığı Risk
Kategorisi
Risk Skoru Tanıdan itibaren toplam sağkalım (yıl)
MDS’den AML’ye dönüşene kadar geçen medyan süre (yıl)
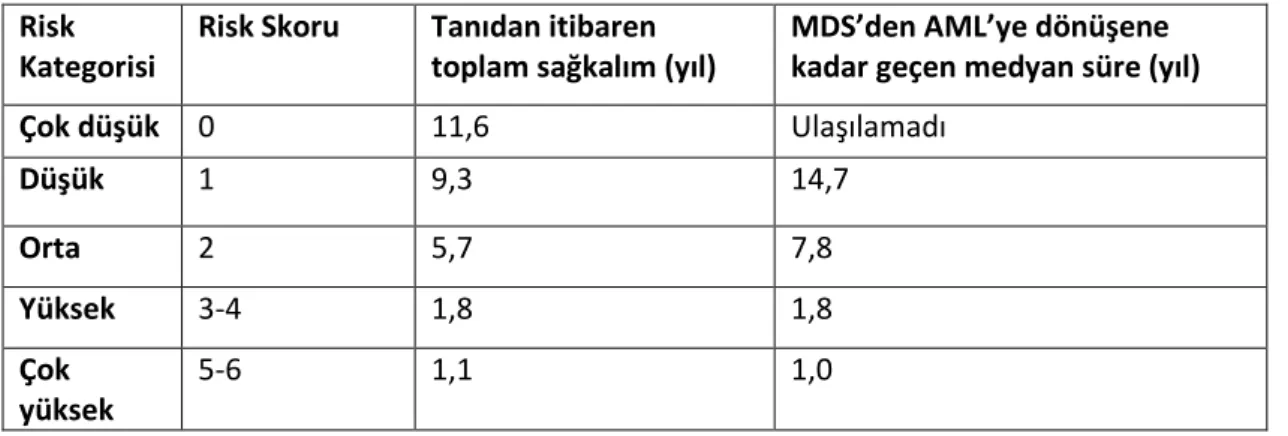
Çok düşük 0 11,6 Ulaşılamadı Düşük 1 9,3 14,7 Orta 2 5,7 7,8 Yüksek 3-4 1,8 1,8 Çok yüksek 5-6 1,1 1,0
WPSS göre genel sağkalım çok düşük risk grubunda;11,6 yıl, düşük risk;9,3 yıl, orta risk5,7 yıl, yüksek risk;1,8 yıl ve çok yüksek risk grubunda 1,1 yıldır. % 25 lösemiye dönüşüme kadar geçen süre sırasıyla;- 14.7, 7.8, 1.8 ve 1 yıldır.
3.2.2 IPSS
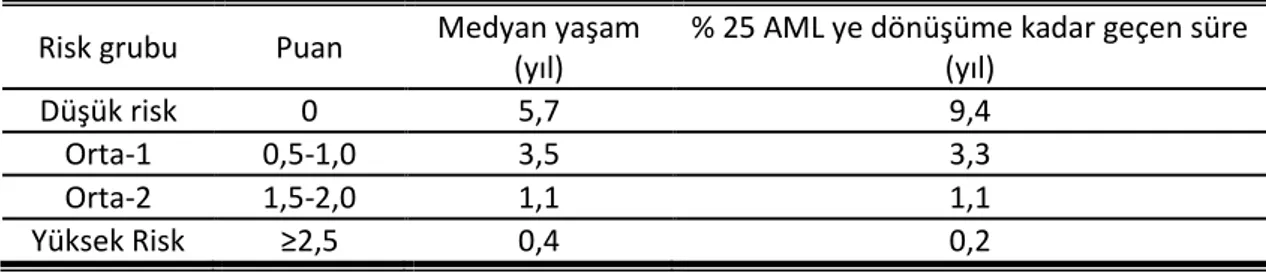
IPSS de kemik iliği blast oranı, karyotip ve sitopeni sayısı kullanılmaktadır. Genel sağkalım sırasıyla; düşük orta-1 orta-2 ve yüksek risk grubu için 5.7, 3.5, 1.2, ve 0.4 yıldır. Yüksek risk grubundaki hastalar AML’ ye dönüşümden ziyade kemik iliği yetmezliğine sekonder nedenlerle kaybedilmektedir.
Tablo 7 IPSS
Puan değeri 0 0,5 1 1,5 2
Kemik iliği blast (%) <5 5-10 11-20 21-30
Karyotip 1 İyi Orta Kötü
Sitopeni 2 0-1 2-3
1
Karyotip
İyi: Normal, -Y, del( 5q), del (20q). Orta: diğer anormalikler
Kötü: Kompleks (≥3 anormallik) veya kromozom 7anomalikleri. 2
Sitopeni Hb<10 gr/dL, MutlakNötrofil Sayısı (MNS) <1.8x109/L, PLT <100x109/L
Tablo 8 IPSS’ ye Göre Genel Sağkalım Ve Lösemiye Dönüşüm Olasılığı
Risk grubu Puan Medyan yaşam
(yıl)
% 25 AML ye dönüşüme kadar geçen süre (yıl)
Düşük risk 0 5,7 9,4
Orta-1 0,5-1,0 3,5 3,3
Orta-2 1,5-2,0 1,1 1,1
Yüksek Risk ≥2,5 0,4 0,2
AML’ye dönüşüm oranları ve lösemiye dönüşüme kadar geçen süre sırasıyla; düşük riskli grupta %31 ve 9.4 yıl; orta 1 grubunda % 39 ve 3.3 yıl; orta 2 grubunda %22 ve 1.1 yıl; ve yüksek riskli grupta % 8 ve 0.2 yıldır.
3.2.3 R-IPSS
IPSS süreç içinde yenilenmiş ve sitogenetik belirteçlerin prognostik önemi daha detaylı dikkate alınmıştır. Oluşturulan bu yeni prognostik sisteme “revised IPSS (R-IPSS)” ismi verilmiştir RIPSS’de sitogenetik, blast, hemoglobin trombosit ve MNS değerlendirilmektedir. Blast sayısı ağırlık kazanmıştır ve sitogenetik 5 gruba ayrılmıştır. Tablo 9 R-IPSS
Prognostik değişken/skor
0 0,5 1 1,5 2 3 4
Sitogenetik* Çok iyi - İyi - Orta Kötü Çok kötü
Blast (%) < 2 - >2-<5 - 5-10 >10 -
Hb gr/dl >10 - 8-<10 <8 -- - -
PLT 109/L > 100 50-100 <50 - - - -
MNS 109/L >0,8 < 0,8 - - - -
*Sitogenetik;
Çok iyi, -Y, del(11q);
İyi, normal, del(5q), del(12p), del(20q); Orta, trizomi 8, trizomi 19, i(17q); Kötü, del7/7q, inv3, del(3q); Çok kötü, kompleks (>3 anomali)
Tablo 10 R-IPSS Göre Genel Sağkalım ve AML Dönüşüm Olasılığı
Risk Grubu R-IPSS
skoru
Genel
medyan sağkalım (yıl)
%25 AML gelişim yüzdesi (yıl) Çok düşük 1.5 8.8 14.5 Düşük >1.5-3 5.3 10.8 Orta >3-4.5 3.0 3.2 Kötü >4.5-6 1.6 1.4 Çok kötü >6 0.8 0.7
Genel sağkalım sırasıyla; çok düşük, düşük, orta, kötü ve çok kötü risk grubu için 8.8, 5.3, 3.0, 1.6 ve 0,8 yıldır. Ek olarak genel sağkalımı tahmin etmek için yaş da kriterlere eklenebilir, ancak AML gelişimini öngöndürmez. IPSS, FAB sınıflamasına dayanılarak yapılmış ancak WHO sınıflamasında >%20 blast AML olarak adlandırılır. Lösemiye dönüşüme kadar geçen süre sırasıyla 14.5, 10.8, 3.2,1.4 ve 0.7 yıldır.
3.3 MDS’de Yeni Sitogenetik Risk Sınıflaması
Son yayınlanan R-IPSS’de sitogenetiğin önemi artmış ve anormallikler 5 gruba bölünmüştür [32]. Y kromozomu ve 11q delesyonu R-IPSS’yi kötüleştirmeyen sitogenetik anormalliklerdir. Normal karyotip, del 5q ve 20q iyi, 7q, Trizomi 8, 19, 21, ve tüm çift anormallikler orta, 3’lü anormallikler kötü ve 3 üzerindeki çok kompleks anormallikler çok kötü grupta değerlendirilmektedir.
Tablo 11 MDS’de Yeni Sitogenetik Risk Sınıflaması [39]
Çok iyi İyi Orta Kötü Çok Kötü
Tek başına del(11q) -Y Normal Tek başına 7q- +8 17q- +19 +21 diğer tek Tek başına(3)(q21q26) >3 anormallik Tek başına der(1;7) del(5q) del(12p) del(20q) Tüm çift anormallikler 3’lü anormallikler 5q(-) eşlik eden anormallik
-7/7q- ile olan ikili anormallikler Del: Delesyon
3.4 Tedavi
MDS de Allojenik kemik iliği transplantasyonu (AKIT) dışında küratif tedavi seçeneği yoktur. Tedavinin amacı semptomları kontrol altına almak, yaşam kalitesini arttırmak ve yan etkiyi en aza indirmek olmalıdır. Asemptomatik hastada tedavinin uzun dönem sağkalımı artırdığı gösterilememiştir. Tedaviye risk grubuna göre belirlenir. IPSS göre düşük ve orta -1 grubu düşük risk, orta -2 ve yüksek yüksek risk grubunu oluşturur.
3.4.1 Eritropoez uyarıcı ilaçlar
Hastada hemoglobin düzeyine bağlı olmaksızın semptomatik anemi olması veya hb<10 gr/dl olması tedavi endikasyonudur. Hemoglobin düzeyi 10 gr/dl üzerinde ise tedaviye sıklıkla gerek yoktur, tedavide hedeflenen hemoglobin değeri 12 gr/dl’dir. Tedavi yanıtın önceden belirlenebilmesi için hastanın transfüzyon ihtiyacı ve serum Eritropetin (EPO) düzeyinin bilinmesi gereklidir. Tedavi öncesi serum EPO<500 U/L ya da >500 U/L olup ayda birden fazla transfüzyon ihtiyacı olan hastalarda önerilir. Serum EPO düzeyi kan transfüzyonundan 14 gün sonrasında bakılmalıdır. Tedaviye 60.000 ünite hafta üç bölünmüş dozla veya tek seferde başlanıp yanıt alınmazsa en fazla 80.000’e kadar çıkılır, yanıt değerlendirmesi 8. haftada yapılır. Hedefe ulaşıldığında 8 haftada bir olmak üzere haftalık doz azaltarak idame doza 30.000 Ü/hafta düşülür. Darbopoetin (DA) için 150 µg/hafta önerilir zayıf hastalar ve böbrek yetmezliğinde doz 2 haftada bir verilir. EPO tedavisine yanıt alınamayan hastalarda tek başına veya eklenerek Granülosit koloni stimülan faktör (G-CSF) uygulanır. 300 µg veya 1-2 ug/kg dozda 1-3 doza bölünerek sc. uygulanır, idamesi haftada 2 doz uygulanmasıdır, nötrofil sayısı 6-10.000/mm3 geçince kesilir. Nötrofil sayısı 6.000/mm3 değerine ulaşan olgularda G-CSF dozu %50 oranında azaltılmalı, 9.000/mm3 üzerine ulaştığında ilaç kesilmelidir. 16. haftadan sonra yanıtın alınmadığı durumlarda tedavi kesilir ve hipometile edici ajanlara geçilmesi önerilir. MDS-RARS olan olgularda tedaviye EPO/DA+G-CSF birlikte başlanmalı ve 16 hafta süreyle uygulanmalıdır. Yanıt değerlendirmesi 16. haftada yapılmalıdır. Yanıt yoksa tedavi sonlandırılmalıdır.
3.4.2 İmmün supresif İlaçlar
3.4.2.1 Antitimosit Globülin-Siklosporin
İmmünsupresif ilaçlar sadece RA veya RCMD, semptomatik anemi ve transfüzyon bağımlılığı veya aşikar trombositopeni veya sık enfeksiyonu olan, yaş < 60, düşük veya orta 1 IPSS skoru, hiposellüler kemik iliği, blast sayısı < % 5 olan hastalarda önerilmektedir. Anti
timosit globülin (ATG) veya tek başına ya da birlikte sklosporin kullanılabilir. Tedaviye yanıt için 3-9 ay beklenmelidir. ATG kullanımı sırasında ve takip eden altı aylık sürede sulfamethoxazol/trimethoprim, asiklovir ve flukonazol koruması yapılabilir. ATG’nin serum hastalığı riski nedeniyle prednison ile kombine edilmesi daha uygundur.
3.4.2.2 Lenalidomid
Lenalidomid semptomatik anemisi olan, IPSS’ye göre düşük/orta I risk grubundaki MDS hastalarında, tek veya çoklu dizi semptomatik sitopenisi olan izole ve/veya diğer anormalliklerle birlikte 5q delesyonu olan hastalarda önerilir. Ülkemizde endikasyonu olmamakla beraber 5q delesyonu olmayan ve semptomatik anemisi olan, IPSS’e göre düşük/orta I risk grubundaki MDS hastalarında, serum EPO > 500 U/L olan hastalarda düşünülebilir. Kök hücre nakli adayı olan genç hastalarda kök hücre rezervinin korunabilmesi için lenalidomid kullanımından kaçınılmalıdır. Tedavi başlangıcında MNS <500/mm3 ve PLT<25.000/mm3 ise lenalidomid tedavisine başlanmamalıdır. Önerilen tedavi dozu 28 günde bir tekrarlanmak üzere, 21 gün süreyle 10 mg/gün’dür. İlk yanıt değerlendirmesi için en az 4 ay süreyle devam edilmelidir.
3.4.2.3 Talidomid
Konvansiyonel tedavilere cevap vermeyen MDS hastalarında kullanım endikasyonu vardır. Talidomid yaygın kullanılmamasına rağmen RA alt tipinde fayda sağlayabilir.
3.4.3 Hipometile Edici İlaçlar
Bu amaçla AZA ve desitabin kullanılmaktadır. 3.4.3.1 Azasitidin
AZA, IPSS sınıflamasına göre orta-II ve yüksek risk grubu hastalarda, nadiren IPSS sınıflamasına göre orta-I olup ağır sitopenileri olan ve standart tedavilere yanıtsız hastalarda kullanılmaktadır. Ayrıca allojenik hematopoetik kök hücre nakli şansı olmayan hastalarda, allojenik hematopoetik kök hücre nakli planlanan hastalarda “köprü” tedavisi olarak, MDS/AML (blast sayısı %20-30) olan hastalarda kullanılabilir. AZA 75 mg/m2 7 gün 28 günde bir en sık kullanılan şemadır. Önerilen kullanım şekli subkutan uygulamadır. Profilaktik önlemlere rağmen subkutan uygulamada lokal ciddi yan etkiler gözlenirse intravenöz (IV) olarak uygulanabilir (Ülkemizde IV uygulama için endikasyon yoktur). Yanıt değerlendirmesi
en erken 4. ayda yapılmalıdır. Dördüncü kür sonu yapılan değerlendirmede yanıt elde edilememiş ancak hastalık stabil seyrediyorsa iki kür daha tedaviye devam edilebilir, hastalık ilerlemesi varsa tedavi sonlandırılır. Hastalıkta ilerlemesi olmadıkça en az altı kür uygulanmadan tedaviye direnç kararı verilmemelidir. Klinik ve laboratuar izlemde ilerlemesi olduğu düşünülen hastalarda ilk dört kürden önce kemik iliği değerlendirmesi ve tedavi değişikliği kararı verilebilir. 4-6 kür sonunda yanıt alınan hastalarda tedavi hastalık ilerlemesine kadar veya tedaviden yanıt gördüğü sürece sürdürülür. En sık görülen yan etki hematolojik toksisitedir. Hastanın klinik durumu göz önüne alınarak düzenli kan sayım takibi yapılmalıdır. Her tedavi öncesi mutlaka kan sayımı yapılmalıdır. Sitopenilerin düzeyine göre doz ayarlaması yapılmalı ancak sitopenilerin primer hastalıkla ilişkisi olduğu düşünülüyorsa doz azaltılmasına gerek yoktur. Ağır sitopenileri ve kötü sitogenetiği olan, artmış blastlı MDS hastalarında sepsis gibi yaşam tehdit edici durumlar dışında doz modifikasyonu önerilmez ve aynı dozda devam edilir. İlk üç siklus sırasında doz azaltmak yerine tedaviyi ertelemek tercih edilebilir. Azasitidin sonrasında ağır nötropenik ateş gelişen hastalarda G-CSF uygulaması düşünülmelidir. Ağır enfeksiyöz bir süreçten geçen hastada bir sonraki siklusta sekonder profilaksi olarak G-CSF ile birlikte kinolon grubu antibiyotik ve/veya antifungal ilaçlar kullanılabilir.
3.4.3.2 Desitabin
IPSS sınıflamasına göre orta-II ve yüksek risk grubu hastalarda, allojenik hematopoetik kök hücre nakli şansı olmayan hastalarda, MDS/AML’de (blast sayısı %20-30) kullanılabilir. 20 mg/m2 IV, bir saatlik infüzyon, 1-5. gün, 28 günde bir tekrarlanır. Desitabin yanıt değerlendirilmesi (kemik iliği biyopsisi) öncesinde aşikar bir progresyon olmadıkça 4-6 kür sonra yapılmalıdır. Hematolojik düzelme olmasa bile hastalığın ilerlemesine kadar tedaviye devam edilir. Hematolojik toparlanma süresi 8 haftayı aşarsa hastalık ilerlemesinin tespiti için kemik iliği değerlendirmesi yapılmalıdır. Hastalık ilerlemesi saptanmazsa, sonraki kür 2 hafta daha geciktirilerek 8 haftada bir sürdürülür. AZA ve desitabinin tedavisi alan hastalar karşılaştırıldığında 65 yaş altı hastalarda sağkalım farkı olmamasına rağmen daha yaşlı hastalarda azasitidinin avantajlı olduğu gösterilmiştir.[40]
3.4.4 Destek Tedavisi
Destek tedavisinde komorbidite varsa hb<10, komorbidite yoksa hb<8 olmadıkça eritrosit süspansiyonu (ES) ve kanama yoksa PLT<10.000 olmadıkça trombosit süspansiyonu önerilmez. Eritrosit transfüzyonunda sadece lökosit filtresi kullanmak yeterlidir, AKIT adayı
hastalarda ES ışınlanmalıdır. Trombosit transfüzyonunun mortalite üzerine etkisi net gösterilmemiştir. Demir şelasyon tedavisi yaşam beklentisi bir yılın üzerinde olan ve transfüzyona bağımlı hastalar, serum ferritin > 1000 mcg/L veya toplam 20 Ü eritrosit transfüzyonu sonrası başlanmalıdır. Deferasiroks (DFX) 10-30 mg/kg/gün dozunda başlanmalıdır. Tedavi etkinliği üçer aylık aralıklarla ferritin düzeyi ile takip edilmelidir. Serum ferritin düzeyinin 500-1000 mcg/L aralığında tutulması hedeflenmelidir, ferritin <500 mcg/L olan hastalarda tedaviye ara verilmelidir. Karaciğer fonksiyon testleri ve kreatinin aylık olarak izlenmelidir. Deferroksamin (DFO) 40 mg/kg ciltaltı infüzyon şeklinde uygulanır. Kısa sürede serum ferritin düzeyi 1500 mcg/L altına düşerse DFO dozu azaltılmalı ve 25 mg/kg üzerine çıkmamalıdır. Enfeksiyonlardan korunmada nötropenik hastalarda rutin antibiyotik profilaksisi önerilmez. Destek tedavisi tüm hastaların tedavisinde en önemli noktadır. 3.4.5 AKİT
Kemik iliği transplantasyonu (KİT), kür sağlayabilen tek tedavi seçeneğidir. Ancak otolog nakillerde yüksek relaps halen önemli bir sorundur. Dolayısıyla Allo-KİT uygun hasta grubunda öncelikle tercih edilen tedavi seçeneğidir IPSS göre orta-2 veya yüksek risk grubundaki MDS’li hastalar ve orta-I risk grubunda olan genç, tedaviye yanıtsız ve semptomatik hastalarda önerilmektedir AKİT; MDS’de kök hücre defektinin ve malign klonun yok edilmesi yolu seçilmiş olgularda % 60'a varan oranda kür sağlayabilir [41]. Ancak MDS hastaların çoğunun 60 yaş üzeri olmasıve mevcut komorbiditeleri nedeniyle bu tedavi modalitesinin MDS’de yaygın olarak kullanımı henüz mümkün görünmemektedir.
3.5 Genetik İnceleme
MDS’de sitogenetiğin önemi skorlama sistemlerinde yer alması ile daha iyi anlaşılmıştır. Hastanın risk grubunun belirlenmesinde ve tedavi kararının verilmesinde öne çıkmıştır. Sitogenetik anormalliklerin saptanmasında karyotip, FISH, CGH, SNP dizileme ve yeni jenarasyon sekanslama yöntemleri kullanılabilir. Standart olarak tüm yeni tanı konulan hastalardan karyotip, yeterli metafaz saptanmayan hastalardan ek olarak FISH istenmelidir. Diğer yöntemler araştırma amaçlı kullanılmaktadır ve gelecekte rutin hale gelebilir.
Tablo 12 Genetik Anormallikleri Saptama Yöntemleri Teknik Duyarlılık Bölünen
Hücre gereksinimi Genel analiz Dengeli transokasyon saptama Uniparental dizomi Anormallik sıklığı Metefaz sitogenetik
Düşük Evet Evet Evet Hayır %50∗
FISH Düşük Hayır Hayır Evet Hayır Değişken†
CGH Yüksek Hayır Evet Hayır Hayır ∼%80
SNP Yüksek Hayır Evet Hayır Evet ∼%80
Yeni jenerasyon
dizileme
Çok yüksek
Hayır Hayır Evet Evet ∼%70¶
∗Sekonder MDS’de yaklaşık % 80 oranında
†Kullanılan panele bağlı aranan lezyonları % 100 oranında saptayabilir. ¶Normal karyotipli bireylerde olmaktadır
3.5.1 Metafaz Sitogenetiği
Konvansiyonel sitogenetik metafazda Giemsa boyası ile bantlama ile kaba ancak yararlı genom araştırması sağlamaktadır. 20 metafaz değerlendirmesinde geniş mutasyonlar ve delesyonlar saptanmaktadır. MDS sınıflamasında karyotip en önemli prognosik faktördür. Karyotip anormalliği (metafaz kromozom analizinde saptanan) primer MDS hastalarının yaklaşık %50’sinde ve hatta daha sık sekonder MDS’de yaklaşık %80 oranında saptanmaktadır [42]. Karyotip anormallikleri MDS’ye spesifik olan monozomi 5 ve 7 olabilir ya da genel olarak bir çok myeloid malignitede saptananan trizomi 8 olabilir. Translokasyonlar birçok hematolojik malignitenin tanı ve prognozunda başlıca dayanaklarından olmasına rağmen MDS’de minör rol oynamaktadır. MDS de en sık kromozomların parsiyel ya da komplet kayıpları izlenmektedir. MDS en yaygın olan yaklaşık % 15 oranında görülen 5q delesyonudur. Daha az yaygın olarak monozomi 7, 7q delesyonu



![Tablo 11 MDS’de Yeni Sitogenetik Risk Sınıflaması [39]](https://thumb-eu.123doks.com/thumbv2/9libnet/3035505.2606/34.892.168.795.349.705/tablo-mds-yeni-sitogenetik-risk-siniflamasi.webp)
![Şekil 5 TET yapısı ve fonksiyonel alanları [75]](https://thumb-eu.123doks.com/thumbv2/9libnet/3035505.2606/51.892.177.701.697.952/sekil-tet-yapisi-fonksiyonel-alanlari.webp)
![Şekil 14 DNA metilasyon ve histon modifikasyon düzenleyici tedaviler [111]](https://thumb-eu.123doks.com/thumbv2/9libnet/3035505.2606/58.892.173.766.514.860/sekil-dna-metilasyon-histon-modifikasyon-duzenleyici-tedaviler.webp)
