T.C
SELÇUK ÜNİVERSİTESİ MERAM TIP FAKÜLTESİ
ÇOCUK SAĞLIĞI VE HASTALIKLARI ANABİLİM DALI
Prof.Dr.Sevim KARAASLAN ANABİLİM DALI BAŞKANI
TURNER SENDROMLU ÇOCUK VE ADÖLESANLARDA SUBKLİNİK ATEROSKLEROZUN
DEĞERLENDİRİLMESİ
UZMANLIK TEZİ Dr. Rıdvan GÜÇLÜ
Tez Danışmanı
Doç.Dr.Mehmet Emre ATABEK
KISALTMALAR AKŞ:Açlık Kan Şekeri AKA: Ana Karotis Arter BBO:Bel Boy Oranı BH:Büyüme Hormonu CRP: C-Reaktif Protein DM: Diyabetes Mellitus
ELAM: Endotelial Lokosit-Adezyon Molekül FGF:Fibroblast Growth Factor
FSH: Folikül Stimulan Hormon
GM-CSF: Granülosit-Makrofaj-Koloni Stimülan Faktör HDL:High Dansite Lipoprotein
HCG:Human Koryonik Gonodotropin
HPLC: High Performance Liquid Chromatography
HOMA-IR: Homeostasis Model Assessment of İnsulin Resistance ICAM: Inter Cellular Adhesion Molecule
IGF: İnsulin-Like Growth Factor İMK: Intima-Media Kalınlığı KAH:Koroner Arter Hastalığı
KA-İMK:Karotis Arter İntima Media Kalınlığı KKH:Koroner Kalp Hastalığı
KVH: Kardiyo Vasküler Hastalık LDL:Low Dansite Lipoprotein Lp(a):Lipoprotein (a)
M-CSF: Macrophage Koloni Stimülan Faktör Mİ: Miyokard İnfarktüsü
MR: Manyetik Rezonans
PDGF: Platelet-derived growth factor SD: Standart Deviasyon
SHOX: Short Stature Homeobox TG:Trigiliserit
TGF:Transforming growth factor TS: Turner sendromu
TK: Total kolesterol TNF :Tumor necrosis factor USG: Ultrasonografi VA:Vucut Ağırlığı
VCAM: Vascular Cell Adhesion Molecule VKİ: Vucut Kitle İndeksi
VLA :Very late antigen
İ
ÇİNDEKİLER
Sayfa numarası1.GİRİŞ……….
42.GENEL BİLGİLER……….
5 2.1. TURNER SENDROMU...5 2.1.1. TARİHÇESi...5 2.1.2. TANIM………...5 2.1.3. EPİDEMİYOLOJİ……….…...6 2.1.4. SİTOGENETİK………....62.1.5. TURNER SENDROMUNUN GENEL ÖZELLİKLERİ……….….7
2.1.6. TANI………..16
2.1.7. PRENATAL TANI………..16
2.1.8. AYIRICI TANI………..17
2.1.9. TURNER SENDROMUNDA TEDAVİ………..18
2.2. ATEROSKLEROZ………..20
2.2.1. ATEROSKLEROZA GENEL BAKIŞ………..…20
2.2.2. ATEROSKLEROZ TANISINDA B-MOD ULTRASON………27
2.2.3. ERKEN TANIDA B-mod USG ………...28
3. MATERYAL VE METOD………....
344. BULGULAR………..….
365. TARTIŞMA………....
446. ÖZET ……….
507. SUMMARY……….
518. KAYNAKLAR………
521. GİRİŞ
Turner sendromu X kromozomunun parsiyel veya komplet yokluğuyla birlikte sıklıkla hücre mozaisizminin eşlik ettiği, karakteristik fiziksel özelliklerin kombinasyonu olarak tanımlanabilir.
Turner sendromunun en sık klinik özellikleri kısa boy ve gonadal disgenezi ile birlikte dismorfik bulgulardır. Fibrotik overler, buna bağlı seksüel gelişim sorunları ve büyüme geriliği sendromun vazgeçilmez iki öğesidir. Yele boyun, kubitus valgus gibi klasik bulgular yanında çok değişik somatik bulgular da görülebilir.
Turner sendromunda mortaliteyi artıran ana neden kardiyovasküler sistem komplikasyonlarıdır ve yaşam beklentisini 13 yaşa kadar indirebilir. Aort koartasyonu ve biküspit aort kapağı en sık görülen yapısal malformasyonlardır. Kardiyovasküler risk faktörlerinin varlığı dikkate alındığında Turner sendromlu hastalarda aterosklerozun erken yaşlarda başlama olasılığı artmaktadır. Aorta kökünün genişlemesi, hipertansiyon ve biküspit aortik kapak aort diseksiyonuna yol açan en önemli nedendir. Oluşacak poststenotik türbülansa bağlı olarak çıkan aorta dilatasyona uğrayabilir ve zaman içinde bu hasara bağlı aort anevrizması ve erken ateroskleroz meydana gelebilir. İskemik kalp hastalıklarına ait ölüm Turner sendromlu hastalarda 7 kat yüksek bulunmuştur. Hipertansiyon, diabetes mellitus, dislipidemi, obezite ve östrojen eksikliği Turner sendromunda iskemik kalp hastalığının riskini arttıran faktörlerdir.Turner sendromlu hastalardaki konjenital kalp hastalığı prevalansı yüzde 17 ile 45 arasında değişir ve belli bir fenotip-genotip ilişkisi göstermez.
Arter görüntülemesinde son gelişmeler ateroskleroz çalışmalarına yeni bir boyut kazandırmıştır. Erken ateroskleroz B-mod ultrasonografi ile periferik arterlerden non invaziv olarak değerlendirilmektedir. Bu teknikle belirgin plak ya da stenoz olmaksızın arteryel intima ve medianın kombine kalınlığı hassas olarak ölçülebilmektedir.
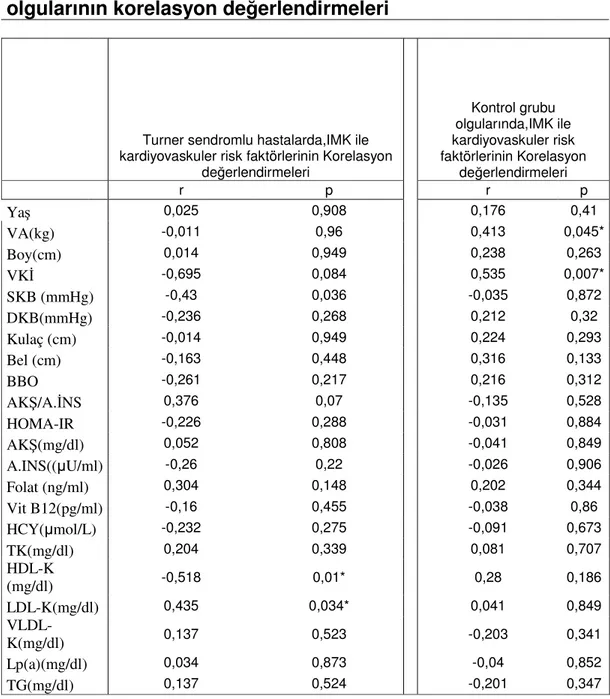
Bu çalışmada Turner sendromlu çocuk ve adölesanlarda subklinik aterosklerozu B mod USG ile karotis arter intima media kalınlığını ölçerek değerlendirdik. Ayrıca KA IMK ile kardiyovasküler risk faktörleri arasındaki ilişkiyi araştırdık.
AO
2. GENEL BİLGİLER 2.1. TURNER SENDROMU 2.1.1. TARİHÇESİ
Turner ve arkadaşları 1938’de ilk kez yedi erişkin kadında seksüel infantilizim, yele boyun ve kubitus valgus birlikteliğini içeren farklı bir antite tanımladılar. Albright 1942’de bu sendromun artmış idrar gonadotropin titresiyle birlikte olduğunu bildirmiştir. 1944’ de Wilkins ve arkadaşları sendromda gonadların yokluğuyla ve olgunlaşmamış ancak gelişimi normal müller kanallarının varlığıyla karakterize patolojik durumu tanımlamışlardır. Ford ve arkadaşları 1959’da Turner sendromlu hastaların seks kromozomlarından birinin eksik olduğunu ve XO cinsiyet kromozomu konfigürasyonu gösterdiğini bildirmişlerdir. Bu açıklamalar sendroma doyurucu açıklama getiren yapıtaşlarını oluşturmuştur(1). Başlangıçta, sadece 45XO vakaları bildirilirken sitogenetikteki ilerlemeler sayesinde, mozaisizm ve yapısal X kromozomu anomalilerine sahip Turner vakaları da bildirilmiş ve sendromun sadece monozomi X olmadığı anlaşılmıştır. Yine aynı ilerlemeler sayesinde, gonadal neoplazi saptanan Turner sendromlu hastalarda Y kromozomu veya Y kromozomuna ait parçacıklar olduğu ve gonadal neoplaziyle direkt ilişkili olduğu gösterilmiştir(2,3).
2.1.2. TANIM: Turner sendromu, fenotipik bulguların yanısıra, parsiyel veya komplet X kromozomu monozomisi ile karakterize bir kromozomal bozukluktur. Bu sendromda bir çok organ ve sistem değişik derecelerde etkilenmesine rağmen, boy kısalığı ve gonadal disgenezise bağlı hipogonadizim en sık karşılaşılan bulgulardır. Yaşa göre boyu 3. persentilin altında veya ortalamanın 2SD altında olan tüm kız çocuklarında Turner stigmatları bulunmasa dahi karyotip tayini yapılarak Turner Sendromu ekarte edilmelidir(4,5).
2.1.3. EPİDEMİYOLOJİ
Görülme sıklığı yaklaşık olarak 1500 ile 2500 canlı kız doğumunda bir dir(2,6). Turner sendromlu fetüslerin yaklaşık %98- %99’u spontan düşükle sonlanmaktadır. Spontan düşüklerin yaklaşık %20’sinin Turner sendromlu fetuslar olduğu bilinmektedir. Ancak düşük eğilimindeki artışın mekanizması henüz aydınlatılamamıştır(2). Hall ve arkadaşları hastaların 1/3’üne yenidoğan döneminde 1/3’üne çocukluk döneminde tanı konulduğunu, geri kalan 1/3’üne ise ancak onlu yaşlarda puberte gecikmesi nedeniyle tanı konulduğunu bildirmişlerdir(7).
2.1.4. SİTOGENETİK
Turner sendromlu hastaların yaklaşık yarısı 45XO karyotipine sahiptir. Diğer yarısında X kromozomunun yapısal anomalileri veya mozaisizm görülmektedir (tablo- 1). 45XO karyotipinde X’ lerden biri tümüyle kaybolmuştur. Klinik bulguların en ağır olduğu durumdur. Bu vakalarda yüz anomalileri, yele boyun ve aort koartasyonu sık görülür. Tüm Turner sendromlular içinde en kısa boylu olanlardır(8).
Yapısal X anomalileri: 46,X,i(Xq), en sık görülen yapısal X kromozomu anomalisidir. Bu durumda X’lerden birinin kısa kolu yoktur ve uzun kolu ise üç kopyadır. Bu hastalar fenotipik olarak 45XO grubuna çok benzerler. 46,X,del(Xp)’de kısa kolun bir kısmı veya tümü kaybolmuştur. Hemen tüm vakalarda fibröz overler, boy kısalığı ve öteki Turner stigmatları vardır. 46, X, del (Xq), uzun kolun delesyonudur. Oldukça nadir görülür. Hastaların çoğunda primer amenore vardır, ancak 45XO’lara oranla menstruasyon oluşan vakalar daha fazladır. Çünkü gonad gelişimi için X’in uzun kolunun tümü gerekli değildir. Boyla ilgili genler daha çok kısa kolunda olduğundan hastaların boyu 45XO’lara oranla daha uzundur ama yine de normalden kısadırlar.
Mozaisizm: Mitozdaki nondisjunction veya anafaz gecikmesinden, iki veya daha fazla hücre serisi oluşabilir. Bu olay, mozaisizm adını alır. 45,X/46,XX TS’de en sık görülen mozaisizm formudur. Mozaik TS’li hastalarda menstruasyon ve meme gelişimi şansı 45XO’lardan daha fazladır. Somatik anomalilerin belirme olasılığı daha düşüktür ve erişkin ortalama boyu 45XO’ lara göre daha fazladır (8,9). TS’ li vakaların %5’inde 45,X/46, XY ve 45X/47XYY mozaisizmi bildirilmektedir. Y kromozomu veya Y
kromozomuna ait fragmanlar taşıyan bu hastalarda gonadal neoplazi görülme sıklığında %30’a varan artma bildirildiği için takipleri önemlidir(3).
Tablo-1. Turner sendromunda rastlanabilen karyotip çeşitleri
Mozaik olmayan karyotipler Tanımlama 45,X XO
46,X i(Xq) X’in uzun kol izokromozomu 46,Xdel (Xp) veya 46XXp X’in kısa kol delesyonu 46,Xdel(Xq) veya 46XXq X’in uzun kol delesyonu 46,Xr(X) Halka X
46,X i(Xp) X’in kısa kol izokromozomu 46,Xi(Yq) Y’nin uzun kol izokromozomu 46,Xt(X;X) XX translokasyonu 46X,t(X;Y) XY translokasyonu Mozaik tipler 45,X/46,XX 45,X/47,XXX 45,X/46,XX/47,XXX 45,X/ 46,XY
2.1.5. TURNER SENDROMUNUN GENEL ÖZELLİKLERİ
Turner sendromunda fenotipik özellikler geniş bir spektrum gösterebilmektedir. Özellikle 45 XO karyotipi taşıyan olgularda fenotip daha belirgindir. Tablo 2 de Turner sendromunda görülen majör klinik özellikler ve görülme yüzdeleri etkilenen sistemlere göre gösterilmiştir(10,11).
Tablo 2. Turner sendromunda görülen majör klinik özellikler
İskelet Sistemi Yüzde(%)
Boy kısalığı 100
Anormal üst / alt oranı 97
Yüksek kubbe damak 71
Karakteristik yüz görünümü (mikrognati ) 60
Kubitus valgus 47 Kısa boyun 40 Kısa 4. metakarp 37 Genu valgum 35 Madelung deformitesi 13 Skolyoz 10 Lenfatik obstruksiyon Kulaklarda rotasyon 63
Düşük ense saç çizgisi 42
Yele boyun 25
El ve ayaklarda ödem 22
Displastik tırnaklar 13
Germ hücre defektleri İnfertilite 99
Gonadal yetmezlik 96
Diğer Sensöri nöral işitme kaybı 50-90 Kardiyo- vasküler anormallikler 55
Multipl pigmente nevüsler 44
Reno vasküler anomaliler 39
Strabismus 18 Pitozis 11 Hipertansiyon 7 Birliktelikler Karbonhidrat intoleransı 4 0 Haşimato tiroiditi 34 Çölyak hastalığı 6
Turner sendromunda klinik özellikler yaşa göre farklılık gösterdiğinden dolayı değerlendirme yapılırken kronolojik yaş dikkate alınmalıdır. Aşağıda belirtilen durumlardan herhangi birisinin bir kız çocuğunda saptanması durumunda Turner sendromunu ekarte etmek için karyotip analizi yapılmalıdır(11,12).
Yenidoğan ve bebeklik dönemi : El ve ayaklarda ödem, ensede katlantı, sol kalp anomalileri ( bikuspid aortik valv, aort koartasyonu, hipoplastik sol kalp ) düşük ense saç çizgisi, düşük kulak, pitozis ve küçük mandibula görülür(13). Yenidoğan döneminde FSH düzeyinin 40U/ L’ nin üzerinde saptanması Turner sendromu açısından dikkatle değerlendirilmelidir.
Çocukluk dönemi
:
Büyüme hızında azalma ile birlikte boy kısalığı, yaşa göre büyüme hızının 10. persentilin altına düşmesi ve bazal FSH düzeyinin belirgin olarak yüksek bulunması, sık otitis media geçirmesi, işitme kaybı, kubitus valgus, hiperkonveks hipoplazik tırnaklar, multipl pigmente nevüsler, 4. metakarp kısalığı, yüksek kubbe damak ve içe dönük gögüs başları görülür(5,13).Pubertal dönem : Yüksek bazal FSH düzeyi ile birlikte 13 yaşına ulaşılmasına rağmen gögüs gelişiminin başlamaması veya atipik gögüs gelişimi ( küçük, belirsiz ve gevşek gögüs uçları ). Yüksek bazal FSH düzeyi ile birlikte primer veya sekonder amenore, duraklamış puberte ve açıklanamayan boy kısalığı Turner sendromunu düşündürmelidir(13) .
Turner sendromunda bir çok organ ve sistem değişik derecelerde etkilenmesine rağmen, boy kısalığı ve gonadal disgenezise bağlı hipogonadizm major komponentlerdir(11,13). Bununla birlikte, Turner sendromu Tablo 2 de verilen klinik özelliklerle birlikte olabildiğinden tanı ve takipte diğer sistemlerin de değerlendirilmesi zorunludur. Puberte gecikmesi olan, özellikle boy kısalığı ile birliktelik olan tüm kızlarda Turner sendromu akla gelmelidir(14).
BÜYÜME
Turner sendromlu bebeklerin doğumlarında ortalama uzunlukları normal aralığın alt sınırındadır. Büyüme hızındaki düşüş 18 aylıkken bile başlayabilir(15). Çoğu çocuk ana okulundaki en kısa çocuk olmayacaktır ancak üçüncü veya dördüncü sınıfta lineer büyüme hızı belirğin derecede düşecektir. Bazılarında hastalık normal pubertal
büyüme sıçramasının gerçekleşmemesiyle anlaşılabilir. Pubertenin gerçekleşmemesi, bu hastaların küçük gözükmesi yanlışlıkla büyüme geriliğine bağlanabilir(16).
Ranke ve arkadaşları Turner sendromunda büyüme sürecinin 4 kategoride incelenebileceğini ileri sürmüşlerdir (17).
1. İntrauterin büyüme geriliği; doğum boyu ve ağırlığı ortalamanın 1 SDS altındadır. 2. Boy gelişimi 0-2 yaş arasında normale yakındır.
3. 2-11 yaş arası belirgin büyüme geriliği; bu dönemde TS ’li hastalar normal kızlara göre 15 cm boy kaybına uğrarlar.
4. 11 yaşından sonra da büyüme yavaştır ancak puberte gecikmesine bağlı olarak büyüme süresi uzadığı için bu dönemde minimal bir boy kaybı olur. Sonuçta total boy uzaması normalin altındadır.
Turner sendromlu vakaların %95’inde boy kısalığı görülür. Hafif intrauterin büyüme geriliği, çocukluk ve adolesan döneminde büyüme yetersizliği ve adölesan dönemde pubertal büyüme sıçraması yokluğu sonucunda bu vakalar kendi toplumları için saptanmış olan ortalama kadın boyundan 15-20 cm kısa kalırlar. Tedavisiz Turner sendromlu vakaların final boyları bir toplumdan diğerine farklılık göstermekle birlikte, 141,9-148 cm arasında değişmektedir(11,18, 19,20,21). Ülkemizde yapılan bir çalışmada tedavisiz Turner sendromu vakalarının final boyu 141,6 cm olarak bulunmuştur. Buna göre ülkemizdeki tedavisiz Turner sendromu vakaları ortalama normal kadın boyuna göre 18,4 cm kısa olarak saptanmıştır(22).
Turner sendromunda boy kısalığı : İntra uterin büyüme geriliği, kemik yapısındaki bozukluklar, SHOX gen mutasyonu, pubertal gelişimin olmaması, BH salımının bozulması, nöro-sekretuar disfonksiyon, BH-IGF aksındaki bozukluklar, IGF’e periferal direnç gibi birden çok faktöre bağlıdır. Turner sendromunda büyümenin değerlendirilmesinde ve takibinde Turner sendromuna özel büyüme eğrileri kullanılmalıdır. Eğer varsa etnik grup ve ülke standartları tercih edilmelidir(4,11).
Brook ve arkadaşları yaptıkları çalışmalarda ebeveyn boyunun hedef boya ulaşmada önemli rol oynadığını gözlemişlerdir. Örneğin uzun boylu ebeveynlerin kızları da uzun boylu olmaktadır(23).
Baş ve boyun anomalileri : Epikantus, göz kapaklarında ptozis, belirgin kulaklar ve mikrognati sık görülen yüz anomalileridir. Görme sorunları, özellikle şaşılık, vakaların yaklaşık %22’sinde görülür. Bazı vakalarda işitme kaybıyla sonlanan kronik süpüratif otitis media görülebilmektedir(24). Katarakt ve nistagmus da sıklıkla görülebiliyor. Kırmızı –yeşil renk körlüğü normal erkeklerle aynı sıklıkta gözükmektedir
(16,25). Östaki kanalının küçük ve disfonksiyone olması ve damak disfonksiyonuna bağlı olarak otitis media, mastoidit ve kolestatom enfeksiyonları erken çocukluk döneminde (1-6 yaş) sıktır. Yaşla birlikte bu enfeksiyonların sıklığı azalır. İletim tipi ve sensöri nöral tip (%50-90) işitme kaybı görülebilir. İşitme kaybı en erken 6 yaşta başlar ve yaş ilerledikçe artar. Bu bakımdan vakalar işitme kaybı bakımından takip edilmelidirler. Strabismus (%18), katarakt, epikantal kıvrım (%46) nistagmus ve pitozis (%13) Turner sendromlu vakalarda sık rastlanan oftalmolojik problemlerdir (4,13,5,11,26).
Süt çocukluğu döneminde ensede yele boyuna sık rastlanır. Embriyonal hayatta boyunda görülen büller ve kistik higromanın zaman içinde değişime uğrayarak bahsedilen yele boyun görünümüne neden olduğu ileri sürülmektedir(27).
Ortodontik problemler :Küçük ve retrognatik mandibula, maloklüzyon ve dental anormalliklere yol açar. Ortodontik muayene 8 yaşından sonra yapılmalıdır(5).
Kardiyovasküler anomaliler
:
Litaratürde Turner sendromlu hastaların %20 ile % 44’ünde kardiyovasküler anomaliler olduğu ve bunlardan aort koartasyonunun % 16 sıklıkla en sık görülen anomali olduğu bildirilmektedir(28). Miller ve arkadaşları ise 35 TS’li hastada yaptığı ekokardiyografi çalışmasında stenozlu veya stenozsuz biküspit aort kapağı sıklığının (%34) aort koartasyonundan daha sık görüldüğünü ileri sürmüşlerdir(28). Ventriküler septal defekt, atrial septal defekt, dekstrokardi ve hipoplastik sol kalp daha az sıklıkla görülen anomalilerdir. Aortik kök dilatasyonu nadir olsa da pubertede ve gebelikte risk teşkil edebilir(29). Hastaların yıllık kardiyak değerlendirilmesinin yapılması tavsiye edilir. Turner sendromunda kardiyovasküler anomalilerin görülme sıklığı % 55’e kadar yükselebilmektedir. Biküspid aortik valv ve aort koartasyonu en sık görülen defektlerdir(4,10). Kardiyak nedenlerle ölüm önemli bir endişe kaynağıdır(16). Aort koartasyonu ve biküspit aort kapağı en sık görülen yapısal malformasyonlardır ondan sonra sol taraflı defektler gelir. Hipertansiyon, mitral kapak prolapsusu ve ileti defektleri de olabilir. Yapısal kalp malformasyonları olmadan ortaya çıkan hipertansiyon genellikle aterosklerotik kalp hastalığı veya böbrek hastalığı ile ilişkili değildir(16). Turner sendromlu hastalarda hiperlipidemi ve koroner arter hastalığı riski net değildir. Turner sendromunda ekokardiyoğrafi tanısal çalışmanın olmazsa olmaz bir parçasıdır, çünkü sadece fizik muayeneyle bikuspid aort kapağını tespit etmek mümkün olmayabilir(30).Böbrek anomalileri
:
At nalı böbrek (%20), çift toplayıcı sistem (%20), böbrek malrotasyonu (%15), tek taraflı böbrek hipoplazisi veya aplazisi görülebilir(31). Turner sendromunda renovasküler anomalilerin sıklığı %30-60’a kadar yükselebilmektedir(13,32). Atnalı böbrek, rotasyonel anormallikler ve çift toplayıcı sistem en sık görülen defektlerdir (11). Hastalarda hipertansiyon, üriner enfeksiyon ve hidronefroza yol açabilirler. Turner sendromunda kalp ve böbrek malformasyonları bulunmadan da hipertansiyon görülebilir(10,33).İskelet anomalileri
: Kemik yaşı adölesan büyüme piki olmadığı zaman 12-14 yaşına
kadar normal sınırlar içinde kalır. Ancak hormon replasmanı yapılmazsa epifizler genellikle 20 yaşına kadar kapanamaz. Kubitus valgus, kısa 4. metakarpaller, tibianın medial kondillerinde deformite, servikal vertebra hipoplazisi ve karpal kemikler arasındaki açılanmada azalma sık görülen iskelet anomalileridir. Kemiklerde osteoporoza benzer görünüm oldukça sık görülür(7).Turner sendromlu vakaların çoğunda uzun kemiklerdeki büyüme vertebral büyümeye göre daha fazla etkilenir ve kronolojik yaş ilerledikçe, orantısız boy kısalığı oluşur. Bazı vakalarda bir veya birkaç servikal vertebradaki hipoplazi, yele boyun deformitesinden bağımsız olarak boyunda kısalığa neden olabilir. Yine mikro ve retrognati, kalkan gögüs, kısa 4. metakarp, madelung deformitesi, kubitus valgus, genu valgum, patellar dislokasyon, konjenital kalça dislokasyonu (%5) ve skolyoz (%10) Turner sendromunda görülen iskelet sistemine ait diğer patolojilerdir. BH tedavisi alan vakalarda skolyoz bulunup bulunmadığına özellikle dikkat edilmelidir (4,10,13,11).
Pubertal dönemde seksuel infantilizim nedeniyle kemik yaşı takvim yaşına göre orta derecede geri kalır. Kemik yaşındaki gerilik uniformite göstermez ve en fazla falangeal kemiklerde bulunurken en az radius, ulna ve metakarpal kemiklerde saptanır (34).
Turner sendromlu çocuklarda yaşa göre kemik mineral dansitesinde azalma bildirilmiştir(11,35). Kemik mineral dansitesi ölçülmeli ve her 3-5 yılda bir tekrarlanmalıdır. Hastalara egzersiz ve özellikle 11 yaşından sonra günlük 1.2-1.5 gr kalsiyum tavsiye edilmelidir(5).
Dermatolojik bulgular
:
Hipoplastik, derin yerleşimli tırnaklar ve aşırı pigmente nevuslar sık görülür. Sebore, hirsuitizm, özellikle ensede olmak üzere deri kıvrımlarında azalma nisbeten sık görülen bulgulardır(36).Turner sendromunda özellikle yüz bölgesine ve yele boyun için uygulanacak elektif plastik cerrahi operasyonlarında keloid gelişebilecegi akılda tutulmalıdır(37). Multıpl melanostik nevüs (%44) görülme sıklığı genel populasyona göre yüksektir(38). Lenfödem
:
Lenfo-juguler bileşke düzeyinde obstriktif malformasyona sekonder olarak gelişir. Spontan rezolusyon gösterebilir. Doğumda el ve ayaklarda görülen ve ilk yıl içinde kaybolan lenf ödem, intrauterin dönemde oluşan ödemin kalıntısıdır. Süt çocuğunda sık olmakla birlikte, herhangi bir yaşta tekrar ortaya çıkabilir. Özellikle büyüme hormonu ve pubertal dönemde östrojen tedavisi ile artabilir. Tedavide sıkı çorap ve diüretikler gerekebilir. Ayrıca, yele boyun, düşük ense saç çizgisi, kulaklarda rotasyonel malformasyonlar, bazı vakalarda görülen meme başı hipoplazisi ve tırnak displazileri gibi malformasyonlar intrauterin dönemde lenf ödemin periferik lenfatikler ve çevreleyen dokular üzerine yaptığı kompresyon süreci sonucunda şekillenmektedir (4,13,33).Neoplaziler
:
45,X/46,XY ile 45,X/47XYY mozaisizmine sahip Turner sendromlu hastalarda gonadoblastom gelişme riski yüksektir. Gonadoblastom görülme yaşı oldukça değişken olup bildirilen en küçük vaka 6 yaşındadır. Yıllık pelvik muayene ve uygun tarama yöntemleriyle gonadal neoplazi bulgularını aramak gerekir. Son zamanlarda geliştirilen moleküler genetik yöntemlerle Y kromozomuna ait DNA daha duyarlı olarak saptanmaya başlanmıştır(39,40).Bu hastalarda eksojen tedavi olmaksızın meme veya seksüel kıl gelişimi olursa bir gonadoblastom veya disgerminom akla gelmeli ve ekarte edilmelidir(41). Y materyali varlığında tümör gelişme ihtimalini ortadan kaldırmak için streak gonadlar puberteden önce çıkarılmalıdır(42,43).
Turner sendromunda nongonadal tümörler de görülebilir. Wertelecki ve arkadaşları 289 Turner sendromlu hastada nongonadal tümör sıklığını araştırmış ve %2.8 bulmuşlardır. Mide, akciğer, vulva, mesane kanseri en sık görülen maligniteler olup bunların sıklığında genel populasyona göre daha sık görüldüğü bildirilmektedir(3).
Yapılan geniş serili çalışmalarda Turner sendromlu vakalarda bazı özel durumlar dışında genel populasyona göre kanser sıklığında bir artışın olmadığı bildirilmiştir. Uzun süre siklik tedavi verilmeden sadece östrojen tedavisi veya uzamış dietilstilbestrol
tedavisi uygulananlarda endometrial karsinoma sıklığı artmaktadır. Y kromozomu veya Y kromozomu fragmanı taşıyan Turner sendromlu vakalarda gonadal tümör riski nedeniyle profilaktik gonadektomi önerilir(5,11).
Genital sistem: İkinci X kromozomunun olmaması veya defektif olması nedeniyle gonadal gelişim mümkün olmayacak ve bilateral streak(çizgi) gonadlar oluşacaktır. Müller kanalları normal gelişimini tamamlayarak iç genital organlar kadın tipinde ve yine dış genital organlar kadın tipinde oluşacaktır. Fakat östrojen uyarımı olmadığı için dış genital organlar infantil görünümdedir. İntrauterin yaşamda ve yeni doğanda overlerde primordial foliküller bulunursa da daha sonra bunlar dejenere olmakta ve puberte döneminde vakaların çoğunda folikül dokusunun yerini tümüyle fibröz bantlar almaktadır. Yani germ hücre yetmezliği patogenezinde, germ hücresi yapım hatası değil, artmış dejenerasyon bulunur. Bu nedenle bu hastalarda çoğunlukla menstruasyon olmaz ve steril kalırlar. Ancak özellikle mozaik formlar(XO/XX) başta olmak üzere Turner sendromlu hastalarda gonadlardaki hızlı dejenerasyona rağmen puberte döneminde menstruasyon görülebilir ve meme gelişimi olabilir ve hatta literatürde nadir olarak bildirilmiş gebelik olguları da vardır. Kanda östrojen düzeyleri düşük gonadotropinler ise yüksektir. Bu çocuklarda surrenal androjenlerin etkisiyle pubik kıllanma oluşur, ama yeterli meme gelişimi olmaz. Sekonder seks karakterlerinin gelişimi, menstruel siklusun devamı ve östrojen eksikliğini karşılamak üzere dışardan hormon replasmanı yapılması uygundur(6,8,42).
Turner sendromunda overler fetal yaşamın 15. haftasına kadar normal gelişirken daha sonra oositler dejenere olmaya ve kaybolmaya başlar ve vakaların %80-90’ında bir yaşına kadar overler fibrotik band şekline dönmüştür. Bu nedenle sekonder seks karakterleri pubertal dönemde spontan olarak gelişmez. Bununla birlikte vakaların ancak %1’inde menstruasyon görülebilir. Turner sendromunda başarılı gebelik oranı ise %1’den azdır(10,33).
Kromozomal çalışmalar: Turner sendromu tanısı için periferal kan karyotip analizi genellikle yeterlidir. Ancak, klinik olarak kesin Turner sendromu tanısı düşünülen vakalarda normal karyotip elde edilse bile fibroblast çalışmaları yapılmalı, daha fazla hücrede karyotip analiz edilerek Turner sendromu tanısı ekarte edilmelidir (5). Turner sendromu fenotipini taşıyan vakaların sitogenetik olarak %50’si 45 X0, %24’ü mozaik yapıda, %17’si izokromozom Xq, %7’si ring X, %2 sinde Xp delesyonu yapısı vardır (43). Xq delesyonlarında ise fibrotik over bulunmasına rağmen birlikte dismorfik bulgular gözlenmez (13,33).
Hormonal çalışmalar: Turner sendromunda hipotalamo-hipofiz aksının intakt olmasından dolayı gonadal disgenezis, artmış bazal veya uyarılmış FSH ve LH düzeyleri ile kendini gösterir. Yenidoğan döneminde FSH düzeyinin 40 U/L nin üzerinde bulunması Turner sendromu bakımından dikkate alınmalıdır. Daha sonra 10 yaşına kadar gonadotropin düzeyleri normal sınırlarda bulunabilir. 10 yaş üzerinde gonadotropin düzeylerinde yeniden belirgin derecede artma saptanır. Turner sendromunda, özellikle yenidoğan ve adölesan dönemde bazal veya uyarılmış gonadotropin düzeylerinin belirgin olarak yüksek saptanması gonadal yetersizliğin göstergesi olarak kabul edilse de, Turner sendromu tanısı için mutlaka karyotip analizi yapılmalıdır (10,13).
Otoimmun ve endokrin hastalıklar
:
Hipotiroidizm, insüline bağımlı diabetes mellitus, romatoid artrit ve inflamatuar bağırsak hastalıkları Turner sendromunda genel populasyona göre çok sık görülür. Bir çalışmada Turner sendromunda otoimmün kaynaklı hipotiroidi için %20 sıklık verilirken başka bir çalışmada %55´e varan tiroid fonksiyon bozukluğu bildirilmektedir(7,44). Haşimato tiroiditi, Turner sendromlu çocuk ve genç erişkin kadınlarda %50’ye kadar yükselen oranlarda görülebilmektedir(4). Vakaların %15 30’unda primer hipotiroidizim gelişebilir(13,11,45). Bu nedenle başlangıçta ve 1-2 yıl aralıklarla serbest T4 ve TSH ve oto- antikor ölçümü yapılmalıdır. Gerekirse USG yaptırılabilir. Vitiligo, alopesi psöriazis normalden sık görülür (12,13). Turner sendromlu vakalar obezite, insülin direnci, dislipidemi, glukoz intoleransı (%40), ve tip 2 diabet yönünden izlenmelidir. Tip 2 diabet normal populasyondan 2-4 kat sık rastlanır. Özellikle oksandrolon tedavisi bu hastalardaki subklinik insülin direncini artırabilir (12,14,45). Turner sendromu vakaları bazen tip 1 diabetle birlikte olabilir(46).Gastrointestinal sistem
:
İntestinal telenjektazi, hemanjiyomatozis, gibi vasküler anomalilere bağlı hayatı tehdit edici kanamalar görülebilir(11,47). Çölyak hastalığı ve diğer inflamatuar barsak hastalıkları riski genel populasyondan yüksektir(48). Turner sendromlu çocuklarda karaciğer fonksiyon testleri bozulabilir(5).Akademik performans: Yapılan çalışmalarda özellikle el ve göz koordinasyonunda bozulma, motor becerilerde zayıflık, kognitif (bilişsel) fonksiyonlarda ortalamanın altında skor elde edilmiş olmasına rağmen Turner sendromlu vakaların genel zeka yaşları normal sınırlar içerisinde kabul edilmektedir. Adölesan dönemde azalmış benlik saygısı ve depresyona eğilim vardır (12,5,45). Bu durum sosyal izolasyona neden olabilir. Böyle durumlarda psikyatrik destek alınmalıdır.
Diğer somatik anomaliler:Kısa ve kalın boyun şekil bozukluğu yaratıyorsa, plastik cerrahiyle mükemmel kozmetik sonuçlar alınabilir.
Kardiyovasküler anomalilerden, aort koarktasyonunun genellikle süt çocukluğu döneminde düzeltilmesi önerilmektedir. Biküspit aort kapağı durumunda stenoza dikkat edilmelidir. Stenozun derecesi kapak distorsiyona uğrayıp kalsifiye oldukça progresif olarak artacaktır. Oluşacak poststenotik türbülansa bağlı olarak çıkan aorta dilatasyona uğrayabilir ve zaman içinde bu hasara bağlı aort anevrizması ve erken ateroskleroz meydana gelebilir. Bazı uzmanlar bu hastalara yıllık ekokardiyografi ve düzenli kan basıncı ölçümü önermektedirler(13).
Bugün biküspit aort kapaklı hastalara subakut bakteryel endokardit proflaksisi önerilmektedir(13).
Hastaya yaşamındaki kısıtlılıklarına uyum sağlayacağı ve böylece çocuk sahibi olmanın dışında tamamen normal bir yaşam sürebileceği güvencesini vermek amacıyla gerek duyulursa ruhsal değerlendirme ve terapotik danışma yapılması uygun olacaktır(1).
2.1.6. TANI
Klinik bulgular her ne kadar Turner sendromu tanısına yaklaştırsa da kesin tanı ancak kromozom analiziyle konulabilir. Bukkal smear çoğu kez en hızlı ve en ucuz tanısal yöntem olmasına rağmen güvenilirliği çok azdır ve bugün kullanılmamaktadır. Bugün, periferik kan lenfosit kültürü ile elde edilen, metafaz safhasındaki en az 20 hücrede giemsa bantlama yöntemiyle kromozom analizi yapılarak tanı konur. Mozaisizm durumunda en az 30 hücrenin incelenmesi gerekir(6,7). Hormonal incelemede FSH ve LH düzeyi yüksek, kan estrojen düzeyi düşüktür(1).
2.1.7. PRENATAL TANI
Turner sendromunun prenatal tanısı, genellikle başka bir endikasyon için (fetal anomali varlığı, ileri anne yaşı vb.) yapılan fetal ultrasonografi, korion villusu veya amniotik sıvı hücrelerinin sitogenetik analiziyle konur.
Ultrasonografide Turner sendromlu fetusta pek çok anomali görülebilir. Kistik higroma oldukça spesifik bir fetal anomalidir, bununla birlikte diğer kromozomal
hastalıklarla (örneğin trizomi 13, 18 ve 21 ) ve nonkromozomal hastalıklarla da birlikte görülebilir. Kistik higroma saptandığı taktirde sitogenetik analiz yapılmalıdır(27,49).
Anormal üçlü yada dörtlü test (HCG, östriol, alfa fetoprotein, inhibin A) Turner sendromu tanısını düşündürebilir. Prenatal karyotipleme post natal olarak doğrulanmalıdır. Spesifik olmamakla birlikte fetal ultrasonografi ile boyun derisinde katlantı artışı Turner sendromunu düşündürebilir. Yine, aort koartasyonu, sol kalp defektleri, brakisefali, renal malformasyonlar, polihidramnios, oligohidramnios ve intrauterin büyüme geriliği Turner sendromlu olgularda prenatal olarak saptanabilen anormalliklerdir(13,50,51).
2.1.8. AYIRICI TANI
İntrauterin ve yenidoğan döneminde kistik higroma, yine yenidoğan döneminde lenfödem, puberte öncesi çocukluk döneminde boy kısalığı, puberte döneminde amenore ve puberte gecikmesi Turner sendromunu akla getirmelidir. Hastaların 1/3’üne yenidoğan döneminde, geri kalan 2/3’üne boy kısalığı nedeniyle çocukluk döneminde veya ancak onlu yaşlarda puberte gecikmesi nedeniyle başvurduklarında tanı konabilmektedir(7).
Ailevi boy kısalığı, hipotiroidizm, büyüme hormon eksikliği, glukokortikoid fazlalığı, kronik hastalıklara bağlı boy kısalığı, Noonan sendromu, kondrodisplaziler, tipE brakidaktili, multipl pterygium sendromu ve Klippel-Feil anomalisi gibi hastalıklarda da boy kısalığı olduğu için Turner sendromu ile ayırıcı tanıya girebilirler(7).
Amenore veya puberte gecikmesi durumunda pür gonadal disgeneziler, Stein-Leventhal sendromu, primer veya sekonder amenorenin diğer sebepleri ekarte edilmelidir. Lenfödem varlığında Milroy hastalığı, tekrarlayan kolestazla seyreden lenfödem, Hennekam sendromu, intestinal lenfanjiektazi ile giden lenfödem akılda bulundurulmalıdır(6).
Fetal kistik higroma, trizomi 21, trizomi 18, trizomi 22 mozaisizmi, fetal alkol sendromu, 13 ve 18. kromozom delesyonları ve Noonan sendromunda da görülebilir(6).
2.1.9. TURNER SENDROMUNDA TEDAVİ
Turner sendromunda uzun süreli ve multidisipliner bir tedavi yaklaşımı gereklidir. Hipogonadizme, boy kısalığı ve diğer somatik anomalilere ve hastanın psikolojik durumuna yönelik olmak üzere tedavi 3 ana başlıkta toplanabilir.
Hipogonadizm: Overlerin transplantasyonu mümkün oluncaya kadar bu durumun tedavisi yalnızca yerine koyucu tedavi olarak kalacaktır. Hormonal tedaviyle sekonder seks karakterlerinin indüklenmesi gereklidir. Düşük doz etinilestradiole genellikle 12-13 yaşlarında ve 2µg/gün dozda başlanmalıdır. Büyümenin durması ve meme gelişiminin Tanner evre 4-5’e ulaşmasını takiben etinilestradiol yerine düşük doz kombine östrojen-progesteron preparatlarına geçilmeli ve tedaviye ömür boyu devam edilmelidir(1,9). Nadiren spontan menstruasyon görülen ve hatta gebelik bildirilen vakalar da vardır(2). Navot ve arkadaşları 1986 yılında ilk kez over yokluğunda endometrial siklusu uyararak ve dışardan ovum implantasyonu yaparak sağlıklı bir gebelik yapmayı başarmış ve TS’li kadınlara çocuk sahibi olma konusunda yeni ışıklar yakmıştır(52).
Pubertal indüksiyon: Turner sendromunda puberte indüksiyonu veya pubertal devamlılığın sağlanabilmesi için östrojen tedavisi gereklidir. Pubertal indüksiyonu yaparken üç konu gözetilmelidir;
1. Normal veya normale yakın pubertal gelişimi sağlamak 2. Büyüme potansiyelini en uygun şekilde kullanmak 3. Uterus kanseri riskini azaltmak
Puberte indüksiyonu için östrojenin başlama zamanı, dozu, preparatın içeriği ve verilme şekli önem taşımaktadır. Östrojenin başlama yaşı ve dozu için en uygun tedavi şeması henüz oluşturulamamıştır. Östrojen tedavisi BH tedavisi ile koordineli olarak ve hastaya özel şartlara göre planlanmalıdır. Büyümenin artırılması öncelik taşıyorsa östrojen tedavisi geciktirilmelidir. Bu hastalarda 12 yaşından önce östrojen tedavisi başlanmaması, hatta 15 yaşına kadar beklenilmesi önerilmektedir. Turner sendromlu vakalarda büyüme hormonu tedavisi 6-8 yaşlarda başlanabilirse yeterli boy uzaması sağlanacağından pubertal indüksiyon tedavi yaşı erkene alınabilir ve kemik kitle kazanımına pozitif katkı sağlanabilir. Östrojen tedavisi düşük dozda başlanıp tedrici olarak artırılır. Erişkin östrojen dozunun 1/4-1/6’sı ile başlanır ve 6-12 aylık aralıklarla hasta cevabına göre doz artırılır. Sekonder seks karakterinin 1-3 yıllık dönemde erişkin özellik kazanması sağlanır. Genel olarak östrojen praparatı oral yoldan verilir. Ancak hepatik yan etkileri gidermek için trans dermal 17- beta östradiol
kullanılabilir(5,13). Östrojen ile puberte indüksiyonuna başlandıktan yaklaşık 1-2 yıl sonra tedaviye progesteron eklenerek siklik tedaviye geçilir. Progesteron başlamanın başka bir kriteri ise devamlı östrojen alırken ara kanamaların olmasıdır. Progesteron dozu karbonhidrat ve lipid metabolizmasına olumsuz etkileri nedeniyle minimum düzeyde tutulmalıdır(5,33).
Pubertal indüksiyon sağlandıktan sonra verilen östrojen ise sekonder seks karakterinin devamlılığını, osteoporozun önlenmesini, arterioskleroz ve kardiovasküler hastalıkların önlenmesini, dişi kimliğin güçlendirilmesi ve vajinal kuruluğun önlenmesini sağlamaktadır.
Boy kısalığı: İlk tanımlandığı yıldan beri boy kısalığına yönelik çeşitli tedaviler denenmişse de ilk yüz güldürücü sonuçlar sentetik büyüme hormonunun keşfiyle başlamış ve tedavide yeni bir dönem başlamıştır. Rekombinant DNA tekniğiyle sentezlenen büyüme hormonu ilk kez Rosenfeld ve arkadaşları tarafından TS’de kullanılmış ve hastaların bu tedaviyle hedef boylarını geçtikleri gösterilmiştir(53). Takiben bu çalışmayı destekleyen çok sayıda çalışma yapılmıştır. Boyu uzatmaya yönelik tüm tedavilerin amacı hasta boyunu normal populasyona yaklaştırmak olmalıdır. Etnik olarak karışık bir toplumda hedef boy uzun olan gruba göre yapılmalıdır. Tek başına boy hızındaki artış tedavide hedefe ulaşıldığı anlamına gelmez(17).
Gerek kısa süreli gerek uzun süreli çalışmalarda BH’nın TS’li hastalarda büyüme hızında artış yaptığı gösterilmiştir(53). Bu çalışmalarda hastalara BH verilmesiyle hem Turner standartlarına göre hem de tedavi öncesi hesaplanan hedef boya göre anlamlı bir boy artışı sağlanmıştır. Doz cevap eğrisi BH eksikliği olanlardan farklıdır ve aynı artışı sağlamak için gereksinim duyulan doz daha fazladır(23). TS’de BH tedavisiyle kemik yaşında belirgin bir artış olmadan büyüme hızında önemli bir artış olduğu ve nihai boyda pozitif etkisi olduğu çeşitli yayınlarda bildirilmiştir. Fakat tedavinin başlangıç yaşı ve optimum dozu arasında farklı görüşler vardır. Genel olarak 0.6-0.7 ü/kg /hafta kullanılarak büyüme hızı, tedavinin ilk yılında iki katına çıkarılabilmekte ve sonra hormonun etkisi takip eden sürede progresif olarak azalmaktadır. Günde bir kez ve subkutan injeksiyon şeklinde kullanılmaktadır(23,53, 54).
Tedavide kullanılan bir diğer ilaç anabolik steroid grubundan olan oxandrolondur. Biyosentetik BH’nın bulunmasından önce tek başına yaygın olarak kullanılmış olan oxandrolonun ilk 2-3 yıllık bir süre için büyüme hızını belirgin olarak artırdığı ancak uzun vadede ulaşılacak nihai boyda önemli bir değişiklik yapmadığı pek çok çalışmada gösterilmiştir(55).
Stahnke ve arkadaşları 212 Turner sendromlu hastayla yaptığı çalışmada, BH’nın tek başına 28ü/m²/hafta verilmesi ile oxandrolon ve 18ü/m²/hafta BH’nın kombine verilmesinin de hedef boyda aynı uzamayı yaptığını bildirmiş ve kombine kullanımı önermişlerdir(55).
Etinilestradiolün büyüme tedavisinde kullanımı hakkında farklı görüşler vardır. Düşük doz(2µg/gün) etinilestradiolün, BH ve oxandrolonla birlikte kullanımını öneren çalışmacıların yanı sıra bazı çalışmacılar hedef boyu etkilemediğini hatta epifizlerin erken kapanmasına neden olarak hedef boyu azaltabileceğini iddia etmektedirler(9,56,57). Örneğin Lyon ve arkadaşları östrojen tedavisinin büyümeyi ilk etapta hızlandırdığını fakat ulaşılacak hedef boyda önemli bir etki yapmadığını vurgulamışlardır(58).
BH tedavisi ve fizyolojiye uygun olarak replase edilen seks steroidleri ile puberte induksiyonu sonucunda Turner sendromlu hastaların erişkin boyunda 5-16 cm lik kazanımlar bildirilmektedir. Bunun sonucunda Turner sendromlu hastaların boyları normal değerlere önemli ölçüde yaklaşmaktadır(13).
Turner sendromunda büyüme hormonu tedavisi bir replasman tedavisi değil farmakolojik etkiden yararlanma tedavisidir. Bu nedenle bu olgulara büyüme hormonu başlanması için uyarı testi yapılmasına gerek görülmediği otoritelerce kabul edilmektedir. BH mümkün olduğunca erken yaşta başlanmalıdır (en erken 3 yaş ) ve 0,045 mg/kg/gün dozuyla başlayıp gerekirse artırılmalıdır. Büyüme 3-6 ay aralıklarla izlenerek kişisel doz ayarlanması yapılamalıdır. Kemik yaşı 13’ü geçince büyüme hormonu tedavisi kesilir. Kemik yaşı 13’ü aşmış hastalarda büyüme hormonu tedavisi başlanmaz. 9-12 yaştan büyük ve büyüme hormonu tedavisi almış ancak halen 3. persentil altında seyreden hastalarda non- aromatizabl(östrojene çevrilmeyen ) oksandrolon tedaviye eklenebilir. Bu ajanlar büyümeyi hızlandırmak için tek başına kullanılmamalıdır. Yüksek dozda kullanıldığında virilizasyon (kliteromegali), glukoz intoleransı ve kemik maturasyonunda hızlanma görülür(13).
2.2. ATEROSKLEROZ
2.2.1. ATEROSKLEROZA GENEL BAKIŞ
Ateroskleroz, erişkinlerde en önemli mortalite ve morbidite nedenidir ve belli bir genetik alt yapı ve riske sahip kişilerde çevresel risk faktörlerinin etkisiyle ortaya
çıkar(59). Ateroskleroz, bütün orta boy ve küçük arterleri tutabilir, gelişimi çocukluk yaşlarından itibaren başlar, ancak erken dönemde damar lümenini tıkamadığı için bulgu vermez. Klinik bulgular, aterosklerotik plak iyice gelişip komplike hale geldikten sonra, erkekte 40, kadında 50’li yaşlardan sonra ortaya çıkar(60).
Ateroskleroz, damar lümeninin değil, duvarının hastalığıdır ve inflamatuvar bir hastalıktır(61). İnflamasyon süreci, genetik kontrol altındadır. Aterogenez, arter endotelinin zedelenmesi ile başlar. Endotel, damar iç yüzeyinde tek sıra halindedir, kana geçirgen değildir, pasif bir bariyer olmayıp son derece aktiftir; endokrin, parakrin, otokrin fonksiyonları vardır ve hemostaz ile vasküler fonksiyonların ayarlanmasında başrölü oynar(62,63). Normalde sağlıklı endotel, kaygan, parlak yüzeyli, vazodilatasyona eğilimli bir yapıdır. Ancak bütün risk faktörleri, oksidatif stres, mekanik, hemodinamik ve şimik etkiler sonucu endotel yapısını bozar(64,65).
Çevresel ve genetik faktörlerin etkisi ile oluşan ateroskleroz gelişiminde üç evre vardır :
1-Yağlı çizgi (fatty streak): En erken ateroskleroz lezyonu olup makroskopik olarak damar yüzeyinden hafif kabarık çizgilerdir ancak lümende tıkanıklığa yol açmazlar.
2-Fibröz plak: Makroskopik olarak görülebilen ve damar lümeninde kısmen obstrüksiyona yol açan yapılardır.
3-Komplike lezyon : Plak fissüre veya rüptüre olduğunda üzerine trombüs oluşumu, plak üzerindeki endotel erezyonuna ya da fibröz kapsülün yırtılmasına bağlıdır. Oluşan trombus, intramüral veya intralüminaldir(60).
Yağlı çizgi gelişimi
Bütün risk faktörlerinin ana etkisini vasküler endotel üzerine yaptıkları sanılmaktadır. Vasküler endotel, bütün damarlarımızın içini kaplayan tek sıra halinde ve kana geçirgen olmayan bir tabakadan oluşur. Endotel hücreleri pasif bir bariyer değildir. Son derece aktif bir organ olup damar tonusu ve pıhtılaşmaya eğilim gibi fonksiyonlarda düzenleyici rol oynar. Normalde sağlıklı endotel, kaygan, parlak yüzeyli, vazodilatasyona egilimi olan bir yapıdır. Ancak yukarıda sayılan risk faktörleri ile karşılaşınca özellik değiştirir. Yapılan çalışmalar, hiperkolesterolemi, hiperhomosisteinemi, sigara yan ürünleri, biyomekanik güçlerde değişim ve hatta bazı infeksiyonların endotel özelliklerini değiştirdiğini göstermiştir. Bütün bu dış etkenler, endotel hücrelerinde bazı genleri aktive ederek bir inflamatuvar süreç başlatır(65). Endotel hücrelerinden bazı adhezyon molekülleri, sitokinler ve büyüme faktörleri salınmaya başlar. Adhezyon moleküllerinden VCAM-1, ICAM-1, ELAM-1 ve
selektinlerin ekspresyonları artar. Büyüme faktörlerinden PDGF, FGF, TGF, IL-1, TNF-ά yanısıra M-CSF, GM-CSF’de salınır. Bu salınan maddelerin etkisiyle, mononukleer hücreler endotele yapışırlar. Mononükleer hücrelerin üzerindeki VLA-4 integrin VCAM-1’e bağlanınca adhezyon sağlanır. Özellikle, okside LDL ile yüklü monositler subendotelyal bölgeye geçerek aktive lipidden zengin makrofajları oluştururlar. Aterosklerozun ilk lezyonu bu köpük hücrelerinden zengin yapı olup yağlı çizgi-fatty streak adını alır. Bu erken lezyon, on yaşındaki çocukların otopsilerinde bile gösterilmiştir. Deney hayvanlarına lipidden zengin diyet uygulandığında da ilk oluşan lezyon yağlı çizgidir(65).
Fibröz plak gelişimi
Zaman içinde risk faktörlerinin de devam etmesi ile bu subendotelyal depolanma giderek artar. Kolesterolden zengin hale gelip büyüyen erken lezyona düz kas hücreleri de göç etmeye başlar. Düz kas hücreleri giderek artan bir ekstraselüler matriks sentezlerler. Bu matriks ve artan düz kas hücre ve lipid depolanması ile lezyon giderek büyür. Arter lümenini kısmen tıkamaya ve klinik olarak semptomların gelişmesine yol açmaya başlar. Ama asıl klinik olaylar bu aterosklerotik plağın rüptüre olması ile oluşur(65).
Komplike lezyon gelişimi
Aterosklerotik plak bir yandan dıştan mekanik stres ve risk faktörlerinin devam etmesi ile yıpranırken, öte yandan içten de yıpranır. Devam eden inflamatuvar süreç nedeniyle plak içi makrofajlar bazı matriks metaloproteinazları salarlar. Bu metallo proteinazlardan örneğin kollejenaz ve diğerleri plağın fibröz çatısını giderek yıpratırlar. Plağın fissüre veya rüptüre olması ile de klinik olaylar ortaya çıkar. Plağın üstündeki endotel ayrılınca, subendotelyal doku kan ile temasa geçer. Subendotelyal doku, faktör VII ve lipoprotein (a) dan zengin olup trombojenik özelliktedir. Bu şekilde trombus oluşumu için bir uyarı ve trombus gelişimi olur. Oluşan trombusun damar lümenini parsiyel veya tam olarak tıkaması ile de akut koroner sendromlar meydana gelir.
Aterosklerotik lezyonların klinik olaylara yol açma potansiyeli plak içeriğine bağlıdır. Eğer plak lipidden zengin, inflamatuvar kompenenti fazla ve düz kas çatısı ince ise, rüptüre olmaya ve klinik olaylara yol açmaya eğilimi fazla demektir. Yoğun olarak risk faktörlerine maruz kalan kişilerde genelde plak içeriği bu tehlikeli özellikleri taşır. Risk faktör modifikasyonu ile plağı bir miktar stabilize etmenin, yani lipid içeriğini azaltıp inflamatuvar özelliği azaltmanın olası olduğu gösterilmiştir(65).
Son yılarda yapılan çalışmalar, aterosklerozun inflamatuvar bir hastalık olduğunu ve bir inflamasyon göstergesi olan CRP’nin (özellikle high-sensitive CRP) serumda yükselmesinin KKH için bir ön belirleyici olduğunu göstermektedir(66,67).
Aterosklerozda klinik, plak içeriğine bağlıdır(68). Plak içeriği: lipid+fibrotik içerikten oluşur. Plak, ne kadar lipidden zengin, inflamatuvar kompenenti fazla ve düz kas çatısı (kapsülü) ince ise, rüptüre olmaya ve klinik olaylara yol açmaya eğilimi okadar fazla demektir. Hassas plaklar, hemoraji içeren plaklar ve büyük kalsifik nodül içeren plaklar trombüse yatkın plaklardır. Büyük lipid çekirdek (lipidden zenginlik), makrofaj artışı (yoğun inflamasyon ) ve ince fibröz kapsül, hassas plağın başlıca özellikleridir. Hassas plaklar, tromboza karşı hassastırlar(60).
Erişkin yaş grubunda en önemli mortalite ve morbidite nedeni olan ateroskleroz sıklığı giderek artmaktadır. Çağımızın hastalığı olan aterosklerozun nasıl geliştiğine ilişkin çok sayıda bilgimiz ve önleyici önlem olmasına karşın, bu önlemlerin tam uygulanmaması nedeni ile hastalık prevalansı giderek artmaktadır(65).
Ateroskleroz yaygın bir hastalık olup bütün orta boy ve büyük arterleri tutabilir. Gelişimi çocukluk yaşlarından itibaren başlayan bu hastalık erken dönemde damar lümenini tıkamadığı için bulgu vermez. Klinik bulgular plak iyice gelişip komplike hale geldikten sonra erkekte dört, kadında beşinci dekatdan sonra ortaya çıkar(65).
Ateroskleroz için risk faktörleri
1. Yaş : Erkeklerde 45, kadınlarda 55 yaş üzeri olmak tüm çalışmalarda ateroskleroz gelişimi için önemli risk olarak belirmektedir.
2. Cinsiyet : Erkek cinsiyet birçok çalışmada başlı başına bir risk olarak belirmektedir. Ayrıca ilerlemiş aterosklerotik damar hastalığının erkeklerde kadınlardan 10-20 yıl daha erken geliştiği bilinmektedir.
3. Sigara kullanımı : Sigara kullanımı, en önemli düzeltilebilir çevresel etkenlerden biridir. Sigara endotel fonksiyonlarını bozar ve HDL-kolesterol düzeylerini düşürür (69). Protrombotik etkileri arasında da kan fibrinojen konsantrasyonunu arttırmak, trombosit tepkilerini arttırmak ve indüklediği sekonder polisitemi yoluyla kan vizkositesini artırmak vardır.
4. Hiperkolesterolemi : Serum kolesterolü ile aterosklerotik damar hastalığı gelişimi arasında sürekli, dereceli ve kuvvetli bir ilişki olduğu yapılan çok sayıda epidemiyolojik çalışmada gösterilmiştir. Deney hayvanlarında kolesterolden zengin diyet ile beslenme ile ateroskleroz gelişimini sağlamak mümkün olmaktadır. Diyet, safra bağlayıcı reçineler, fibratlar veya niasin tedavisi kullanılarak kolesterolün
düşürülmesiyle koroner olay ve inme sıklığını azaltmanın mümkün olduğu çok sayıda çalışmada kanıtlanmıştır(70).
5. Hipertansiyon : Hipertansiyon, aterogeneze birkaç mekanizma ile katkıda bulunur(71). Endotel disfonksiyonu hipertansiyonun erken evrelerinden itibaren ortaya çıkar. Endotele bağımlı vazodilatatörlere yanıtın azalması, lipoproteinlere damar permeabilitesinin artması, endotelin üretimi ve artmış lökosit yapışabilirliği endotel disfonksiyonunun aterogenezi destekleyen olumsuz etkileridir. Kan basıncı ile kardiyovasküler risk arasındaki ilişkinin devamlı olduğu birçok çalışmada gösterilmiştir. Gerek sistolik, gerek diastolik kan basıncı ile koroner olay ve inme gelişme riski arasında önemli bir ilişki vardır ve tedavi ile bu riski azaltmak mümkündür.
Batı ülkelerinde hipertansiyon prevalansı yüksektir ve yaşla artar. Farklı coğrafyalarda, değişik etnik kökenli kişilerde yapılan araştırmalarda kan basıncı ile KAH ve inme arasında doğrudan bir ilişki saptanmıştır. Dört yüz yirmi bin kişiyi kapsayan dokuz prospektif çalışmanın metaanalizinde, yüksek diyastolik kan basıncı olan kişilerin, normal diyastolik kan basıncı olan kişilere göre KAH riskinin 5-6 kat fazla olduğu bulunmuştur(72). Üç yüz bin kişinin izlendiği bir başka çalışmada da koroner mortalite ile hem sistolik hem diyastolik yüksek kan basınçları arasında ilişki bulunmuş, ancak yüksek sistolik basınçta risk daha yüksek bulunmuştur(73).
6. Diabetes Mellitus : Diabetes Mellitusun da birden fazla mekanizma ile ateroskleroza yol açtığı sanılmaktadır(74). Hipertrigliseridemi ve düşük HDL-K yanı sıra, bazı büyüme faktörleri ve insülinin dolaşımda artmasının aterogenezde rol oynadığı düşünülmektedir. Ayrıca diabetik hastalarda plazminojen aktivatör inhibitör düzeylerinde artış ve tromboza eğilim de vardır. Diabetes mellituslu hastalarda, lipoprotein düzeyleri, örneğin LDL kolesterol düzeyi anormal olmaya bilir. Ancak lipoproteinler glikolize olabilirler ve bu da fonksiyonlarında anormalliklere yol açar. Gerek insüline bağımlı, gerek bağımlı olmayan diyabetin önemli bir koroner risk faktörü olduğunu gösteren çok sayıda epidemiyolojik çalışma vardır.
7. HDL kolesterolün düşük olması : HDL kolesterolün ateroskleroz gelişiminden koruyucu bir rolü vardır (75). Aterosklerotik lezyonlardan kolesterolün geri alınmasının HDL-K tarafından ve muhtemelen reseptör bağlantılı mekanizmalarla sağlandığı sanılmaktadır. Özellikle apoprotein A1 içeren HDL, apoprotein A2 içeren HDL-K ‘ya göre daha fazla koruyuculuk özelliği içerir. Düşük HDL-K, yani 35 miligram desilitre altındaki HDL-K değerlerinin aterosklerotik damar hastalığı gelişimi için önemli ve bağımsız bir risk faktörü olduğu bilinmektedir .
8. Aile öyküsü: Aterosklerotik damar hastalığı gelişiminde en güçlü etmenlerden biri aile öyküsüdür. Ailesinde prematür aterosklerotik damar hastalığı öyküsü olan kişilerde erken koroner ateroskleroz riski 12 kat artar. Bu yatkınlığın bir kısmı genetik temellerin bilinen çeşitli kardiyak risk faktörlerine bağlı olabilir. Bunlar arasında tek gen mutasyonuna bağlı lipid metabolizması bozukluklarından başka, hipertansiyon, diabetes mellitus ve diğer metabolik bozukluklar gibi daha karmaşık poligenik bozukluklar da sayılabilir.
9. Obezite: Framingham çalışmasında hem erkeklerde hem de kadınlarda obezite kardiyovasküler hastalık için bağımsız bir risk faktörü olarak bulunmuştur(65). Beden kitle indeksi ile mortalite arasında doğrusal bir ilişki vardır. Şişmanlık ile hipertansiyon, glikoz intoleransı, trigliserid yüksekliği, HDL-K düşüklüğü sıklıkla birlikte olduğundan koroner risk artışına katkıda bulunurlar. Şişmanlığa bağlı risk yalnız şişmanlığın derecesi ile değil yağın vucuttaki dağılımı ile de ilgilidir. Yağın karın bölgesinde toplanması ile karakterize abdominal obezite KAH riskini özellikle arttırır. Erkeklerde 0.9, kadınlarda 0.8 altındaki bel kalça oranları normal kabul edilir.
10. Hiperhomosisteinemi: Aterosklerotik damar hastalığı için plazma homosistein değerinin 15µmol/L üzerinde olmasının bağımsız risk faktörü olduğu çeşitli çalışmalarda gösterilmiştir(76). Physicians Health Study Çalışmasında homosistein düzeyi bu değerin üzerinde bulunanlarda MI geçirme riski 3.4 kat artmış olarak bulunmuştur(77). Homosistein KAH riskini endotel üzerine zararlı etkileri ve antikoagülan aktivite üzerine olan olumsuz etkileri yoluyla yaptığı düşünülmektedir. Homosistein düzeyinin başlıca belirleyicisi metyonin tetrahidrofolat redüktaz genotipi ile plazma vitamin B12 ve folat düzeyleridir. Diyete vitamin B12 ve folat eklenmesiyle homosistein düzeyini düşürmenin mümkün olduğu bazı çalışmalarda gösterilmiş olmasına rağmen böyle bir girişimin KAH mortalite ve morbiditesini ne ölçüde etkileyeceği konusu ancak sürmekte olan çalışmalar sonuçlanınca ortaya çıkacaktır.
11. Lipoprotein(a): Çeşitli vaka kontrollü çalışmalarda KAH ile lipoprotein (a) düzeyleri arasında bir ilişki olduğu bildirilmiştir. Ancak ileriye dönük olarak yapılan çalışmalarda lipoprotein (a)’nın KAH için risk faktörü olup olmadığı konusunda farklı sonuçlar elde edilmiştir(78). Erkek ve kadınlarda risk ayrı ayrı değerlendirildiğinde de farklı sonuçlar ortaya çıkmıştır. Artmış lipoprotein (a) düzeyi birlikte artmış LDL-K düzeyleride de bulunuyorsa KAH için risk oluşturmaktadır. Lp (a)’nın 30 mg/dL üzerinde olması KAH için risk yaratmakta, ancak Lp (a) üzerine etkisi olmayan ilaçlarla LDL-K düşürüldüğünde Lp (a)’ya bağlı risk azalmaktadır. Tek başına Lp (a)
düzeyini düşürmenin yararını gösteren bir çalışma da bulunmadığından, bugün için rutin Lp (a) ölçümü önerilmez, ölçülüp yüksek bulunan kişilerde de daha çok diğer risk faktörlerinin tedavisine ağırlık verilmelidir.
12. Östrojen eksikliği: Kadınlarda ilerlemiş aterosklerozun erkeklerden daha geç ortaya çıkması hormonal faktörlere bağlandığından, menapoz sonrası östrojen tedavisinin yararlı olabileceği düşüncesiyle bu yönde çeşitli araştırmalar yapılmıştır. Çeşitli gözlemsel çalışmalarda böyle bir tedavinin risk azaltıcı olabileceği yönünde sonuçlar elde edilmiştir (79).
Tablo 3. Ateroskleroz için risk faktörleri Majör risk faktörleri
Sigara içme Hipertansiyon
Serum kolesterol yüksekliği HDL kolesterol düşüklüğü Diabet mellit
Kondisyonel risk faktörleri Trigliserid yüksekliği
Küçük yoğun LDL Lipoprotein(a) Homosistein
Pıhtılaşma faktörleri
Plazminojen aktivatör inhibitör-1 Fibrinojen
C-reaktif protein
Predispozan risk faktörleri
Obezite (özellikle abdominal obezite) Fiziksel aktivite azlığı
Erkek cinsiyet
Ailede erken yaşta KKH bulunması Sosyal ve ekonomik faktörler
Psikolojik faktörler İnsülin direnci
2.2.2.
ATEROSKLEROZ TANISINDA B-MOD ULTRASONYıllardır arter lümenini değerlendirmede kullanılan anjiografi ve Doppler ultrason gibi teknikler arteryel hastalıkların klinik sonuçlarını değerlendirmek için oldukça kullanışlı yöntemlerdir. Anjiografideki gelişmelere rağmen, aterosklerozun erken evresinin tanısı ya da arter lümenini tıkamayan ilerlemiş duvar hastalığında bu teknik yetersiz kalmaktadır. Buna karşın B-mod USG arter duvarı hastalığının erken evresinde güvenilir sonuçlar vermektedir.
Arter görüntülemesinde son gelişmeler ateroskleroz çalışmalarına yeni bir boyut kazandırmıştır. Geçmiş yıllarda anjiografi ve Doppler USG arter lümen ölçümü ve stenoz yüzdesini hesaplamak için kullanılıyordu. Duvar kalınlığı ise lümen stenozundan hesap ediliyordu. Son yıllarda araştırmacılar B-mod USG’yi arter duvarını direkt olarak ölçmek için kullanmaktadır. Anjiografik ve Doppler USG çalışmaları halen obstruktif hastalıkların prognuzunu değerlendirmede kritik öneme sahiptir. Damar duvar ölçümünde kullanılan yeni teknikler ise erken aterosklerozun risk faktörleri ile ilişkisini ve ateroskleroz gelişmesinin değerlendirilmesinde daha kullanışlıdır. Anjiografi invaziv bir yöntem olduğundan, bu nedenle kullanımı semptomatik hastalarla sınırlı kaldığından, populasyon çalışmalarında kullanılamaz. Anjiografik çalışmaların noninvaziv tarama teknikleri sonrası uygulanması önerilmektedir(80).
B-mod USG
Farklı dokularda akustik farklılıkları ve doku sınırlarındaki sinyallerin lokalizasyonun değerlendirilmesine dayanmaktadır. B- mod USG aterosklerotik plakların morfolojisi, semptomatik sonucu ve risk faktörleri ile ateroskleroz arası ilişkide büyük bir olanak sağlamaktadır(81).
Doppler yöntemi ile arterlerde sertleşme ölçülebilmiş ve yaşla birlikte bu değerlerde artış olduğu gösterilmiştir(82). Doppler USG ile belirgin stenoz olmaksızın da aterosklerotik plağın neden olduğu türbülans saptanabilir, ancak akım bozukluğu ileri derecede olmalıdır. Nonstenotik plakların saptanmasında Doppler USG duyarlılığı azdır. Bu nedenle Doppler ve B-mod USG tekniklerinin kombinasyonu daha bilgi verici olacaktır(83). Aterosklerotik plak histolojisi ile ultrasonik görünümü arasında ilişki gösterilmiştir(84). Ateroskleroz takibinde USG ile biyopsi alınması da önerilmektedir (85,86).
2.2.3. ERKEN TANIDA B-mod USG
Ateroslerozun klinik bulgu ve belirtilerinin saptanabilmesi için lezyonların orta ya da ileri evrede olmaları gerekmektedir. Erken evrelerde aterosklerotik lezyonlar arter duvarındaki simultane dilatasyon sonucu lümen çapını azaltmaksızın ilerleyebilirler. Bu da arteriografi ile stenozun tanısını güçleştirmektedir. Halbuki B-mod USG ile belirgin plak ya da stenoz olmaksızın da arteriyal intima ve medianın kombine kalınlığı hassas olarak ölçülebilir. Karotis arterinden ölçüm yapılması geniş populasyon çalışmalarında erken aterosklerozun saptanmasında en doğru yöntem olarak kullanılabilir.
Zweibel ve arkadaşları ilk kez arter duvarının B-mod görüntülemesinde hipoekojenik boşlukla ayrılan birbirine paralel iki ekojenik çizgileri saptamış ve bunları I ve M çizgileri olarak tarif etmiştir. Bu çizgiler intima – media kompleksini oluşturmaktadır. Arter arka duvarının I-M kompleks kalınlığı histolojik olarak intima +media kalınlığına uymaktadır(87). Dallanmayan arterlerde (örnegin arteria karotis komminisin distal 1,0 cm’lik kısmı) intimal kalınlık çok küçük ve ince bir bağ dokusu üzerindeki tek bir tabaka endotel hücrelerinden oluşmaktadır. Buda sadece yaklaşık 0,02 mm olup, mevcut B- mod USG teknikleriyle ayırt edilemez. Böylece ultrasonografik olarak ölçülen KA-İMK’ın çoğunluğu tunika media dan oluşmaktadır.
Pignoli ve arkadaşları ultrasonografik ve histolojik ölçümlerin uyumluluğunu aterosklerotik ve sağlıklı karotis arterlerinde göstermişlerdir(88). Ölçümlerde daha sabit ve tekrarlanabilir olduğu için karotisin arka duvarı kullanılmıştır. Ateroskleroz karotis arterinde genellikle bifurkasyondan başlar ve proksimal olarak arteria karotis komminise doğru ilerler. Arteria karotis internada daha ziyade fokal plak oluşumu görülmekte iken, arteria karotis komministe kalınlaşma diffüz olmakta, bu nedenle ölçümlerde arteria karotis komminis tercih edilmektedir(89). Arterlerdeki intima-media kompleks kalınlığındaki değişiklikler B- mod USG ile görüntülenebilmekte ve bu değişiklikler aterosklerotik lezyonlardan önce oluşabilmektedir .
Epidemiyolojik ve klinik çalışmalarda B-mod USG’nin avantajları, noninvaziv olması, semptomatik olmayanlarda uygulanabilmesi, kolaylıkla tekrarlanabilmesidir. Semptomatik plak gelişiminden önce intima-media kompleksindeki morfolojik değişiklikler değerlendirilerek ateroskleroz gelişimi incelenebilir .
USG ile erken aterosklerozun değerlendirilmesinde farklı yaklaşımlar önerilmiştir. KA-İMK ölçümü kalitatif iken, ultrasonografik biyopsi(UB) morfolojik, kantitatif değerlendirmedir. UB tek bir hastayı değerlendirmek için daha kullanışlı iken,
populasyon çalışmalarında KA-İMK ölçümü daha kolaydır(90). 60-80 yaş arasındaki 1106 olguda yapılan çalışmada KA-İMK ölçümü ile arteria karotis kommunisteki ateroskleroz, erkeklerde kadınlara oranla 5 ile 10 yıl daha ilerlemiş bulundu. KA-İMK yaşla artmakta idi. Olguların periferik arter hastalığına bağlı semptomatik olanlarında KA-İMK daha da artmış idi. Olgularda ciddi ateroskleroz prevalansı herne kadar az olsa da, KA-İMK’ta hafif artışlar bile periferik arterlerde klinik olarak anlamlı aterosklerozla sonuçlandı(91).
Veler ve arkadaşları sağlıklı insanlarda KA-İMK değerinin 0,36 ile 1,07 mm arasında değiştiğini göstermiştir(87). Bir başka çalışmada karotis aterosklerozu KA İMK’ın 1,0 mm’den fazla olması olarak tanımlanmıştır(92).
İntima- media kalınlığı ilk kez 1986 yılında Pignoli tarafından B- mod ultrason ile ölçülmüştür(88). Daha sonraları cerrahi olarak çıkartılan aortada ki İMK‘nın ölçümlere çok yakın olduğu gösterilmiştir(93). 1990’lı yıllarda ölçümlerin daha rahat yapılabilmesi ve de karotis arterinin sık olarak incelenmesinden dolayı İMK ölçümünde karotis arteri kullanılmaya başlanılmıştır(92). O tarihten beri yapılan çeşitli çalışmalarının sonucunda KA-İMK aterosklerozu belirlemede yeni bir parametre olarak kullanılmaya başlanmıştır(93-96). Arterler, en içte intima, orta da media ve en dışta adventisya olmak üzere üç tabakadan oluşurlar(97). İntima tek sıra endotel hücre tabakasından oluşur ve de aterosklerotik lezyonun oluştuğu bölgedir. Media tabakası düz kas hücrelerini, elastik ve kollojen liflerini içerirken, adventisya tabakası en değişken tabaka olup yoğun kollojen ve elastik lifler içermektedir. İMK, intima media kompleksini yani endotel hücrelerini , konnektif dokuyu, düz kas hücrelerini ve de plak oluşumu için gerekli olan lipid yoğunluğunu gösterir(97).
İMK’ nın ultrason ile gösterilmesi intima ile mediayı birbirinden ayıramaz(98). İMK’nın artışı intima ve /veya media tabakasının kalınlaşması sonucunda olmaktadır(98,99). İntimal kalınlaşmadan primer olarak endotel fonksiyon bozukluğu sonucu oluşan ateroskleroz, medianın kalınlaşmasından ise genellikle hipertansiyona bağlı oluşan düz kas hipertrofisi sorumlu tutulmaktadır. Hipertansiyonda gözlenen bu vasküler hipertrofi genellikle sol ventrikül hipertrofisi gelişmeden gözlenen erken ilk bulgudur. Kan basıncının yüksek devam etmesi halinde endotel hasarı oluşarak aterosklerozda meydana gelebileceği gibi, primer olarak endotel fonksiyon bozukluğuna yol açan bir çok faktörde aynı zaman da düz kas hipertrofisine yol açmaktadır.