T.C.
EGE ÜNİVERSİTESİ TIP FAKÜLTESİ
TIBBİ GENETİK ANABİLİM DALI
Bardet Biedl Sendromlu Hastalarda
Moleküler Genetik Analiz İle
Fenotip-Genotip Korelasyonu
Dr. Aslı ECE SOLMAZ
Uzmanlık Tezi
Tez Danışmanı
Prof. Dr. Ferda ÖZKINAY
Ağustos 2014 İzmir
ÖNSÖZ
Hayatımın dönüm noktalarından biri olan, 4 yıl önce başladığım uzmanlık eğitimimi severek hazırladığım bu tez ile sonlandırmaktayım. Hep duyduğum ama çok da bilmediğim Bardet Biedl Sendromu’nu tez konusu olarak öneren sevgili tez hocam Prof. Dr. Ferda Özkınay’a, sendromu derinlemesine incelememe, hastaları yakından tanımama, kliniği sevmeme, yeni nesil dizi analizini öğrenmeme ve en önemlisi ufkumu genişletmeme yardımcı olduğu için çok teşekkür ederim. Bu süreçte silyopatiler olarak sınıflanan ve fark etmesekte tıbbın pek çok alanında karşılaştığımız hastalıklar grubunu daha yakından tanıma fırsatı buldum.
Tez çalışmamda bilgisi, tecrübesi ve çalışma disipliniyle bana yol gösteren Doç. Dr. Hüseyin Onay’a, çalışma sürecinde bana destek olan Uzm. Dr. Ayça Aykut’a ve çalışma sırasında beni yalnız bırakmayan Aşkın Özel’e teşekkür ederim.
Uzmanlık eğitimim süresince bilgi ve tecrübelerinden yararlanığım, her zaman daha iyisini yapmamı isteyen ve beni destekleyen hocalarım, Prof. Dr. Cihangir Özkınay’a, Prof. Dr. Özgür Çoğulu’ya, Doç.Dr. Haluk Akın’a, Yrd. Doç. Dr. Emin Karaca’ya ve Yrd. Doç. Dr. Asude Durmaz’a çok teşekkür ederim.
Bu süreçte bana yardımcı olan Uzm. Dr. Burak Durmaz’a, hastalara bakışımı değiştiren ve klinik becerilerimi arttıran Çocuk Sağlığı ve Hastalıkları rotasyonumda bana yol gösteren ve yardımcı olan Uzm. Dr. Tahir Atik’e teşekkür ederim.
Acısıyla tatlısıyla bu dört yılı birlikte geçirdiğim, pek çok şeyimi paylaştığım, bana destek olan, birlikte çalışmaktan keyif aldım aistan arkadaşlarım, Dr. Esra Ataman’a, Dr. Taha Reşit Özdemir’e, Dr. Merve Saka Güvenç’e, Dr. Biray Ertürk’e, Dr. İsmihan Merve Tekin’e, Dr. Hasan Taşlıdere’ye, Dr. Ayşe Nur Kavasoğlu’na ve Dr. Hilmi Bolat’a teşekkür ederim.
Eğitim sürecim boyunca bana işin püf noktalarını öğreten, yardımcı olan ve dostuk yapan tüm teknisyen, biyolog, sekereter ve personele çok teşekkür ederim.
Beni bugünlere getiren başta anne ve babam olmak üzere sıradışı aileme, beni ben yaptıkları için teşekkür ederim.
Son olarak, hayattaki yol arkadaşım, hem sevgilim hem de en iyi dostum olan eşim Ulaş’a ve adı gibi dalgalı, heyecanlı, duru ve doyulmaz olan oğlum Utku Deniz’e yanımda oldukları için teşekkür ederim.
İ
ÇİNDEKİLER
İÇİNDEKİLER...iii ŞEKİL DİZİNİ... v ÇİZELGE DİZİNİ...vii KISALTMALAR ...viii ÖZET ... xi ABSTRACT ...xii 1. Giriş ... 1 2. Literatür özeti ... 32.1 Silya yapı ve fonksiyonu... 3
2.2 Silyopatiler ... 4
2.3 Bardet Biedl sendromunun tarihçesi ... 5
2.4 Prevalans ... 6
2.5 Bardet Biedl sendromunun klinik özellikleri ... 6
2.5.1 Major bulgular... 8 2.5.1.1 Obezite... 8 2.5.1.2 Rod-kon distrofisi ... 8 2.5.1.3 Polidaktili ... 9 2.5.1.4 Genital anomali... 9 2.5.1.5 Böbrek anomalisi ... 9 2.5.1.6 Öğrenme güçlüğü ... 10 2.5.2 Minör bulgular... 10
2.6 Bardet Biedl sendromunda genetik heterojenite ... 11
2.7 Bardet Biedl sendromunda genotip-fenotip korelasyonu ... 13
3. GEREÇ ve YÖNTEM... 15
3.1 Olgu Seçimi ... 15
3.2 Olgularda Moleküler Genetik Çalışma ... 15
3.2.1 Örneklerin Toplanması ve DNA İzolasyonu ... 16
3.2.2 Araştırılan DNA Bölgelerinin Çoğaltılması ... 16
3.2.3 Mutasyon saptanan gen bölgelerinin doğrulanması ve ebeveynlerdeki taşıyıcılığın belirlenmesi... 23
3.2.4 Amplifiye Edilen Bölgenin Değerlendirilmesi... 25
3.2.5 PCR Ürünlerinin Birinci Pürifikasyon İşlemi ... 26
3.2.7 Ürünlerin İkinci Pürifikasyon İşlemleri ... 28
3.2.8 Örneklerin Sekans Cihazına Yüklenmesi... 29
4. Bulgular... 30 4.1 H.A (Olgu 1) ... 31 4.2 M.Ö (Olgu 2) ... 33 4.3 U.A (Olgu 3) ... 36 4.4 B.D (Olgu 4)... 38 4.5 Z.B (Olgu 5)... 41 4.6 S.Y (Olgu 6) ... 43 4.7 E.Ö (Olgu 7) ... 46 4.8 D.S (Olgu 8) ... 48 4.9 V.Ö (Olgu 9) ... 51 4.10 E.N.Ç (Olgu 10) ... 53 4.11 B.Y (Olgu 11) ... 56 4.12 A.Ö (Olgu 13)... 58 4.13 N.E (Olgu 15) ... 60 5. Tartışma ... 66 KAYNAKLAR ... 93
Ş
EKİL DİZİNİ
Şekil 1. Primer silyanın yapısı ve intraflagellar transportta görevli genler ... 4
Şekil 2. BBS Genlerinin PCR Ürünleri Jel Görüntüsü... 26
Şekil 3. 1 No'lu Olgunun Aile Ağacı... 32
Şekil 4. 1 No'lu Olgunun Klinik Bulguları ... 32
Şekil 5. 1 No’lu Olgunun Sanger (a) ve MiSeq (b) Dizi Analizi Görüntüsü ... 33
Şekil 6. 2 No'lu Olgunun Aile Ağacı... 34
Şekil 7. 2 No'lu Olgunun Klinik Bulguları ... 35
Şekil 8. 2 No’lu Olgunun Sanger (a) ve MiSeq (b) Dizi Analizi Görüntüsü ... 35
Şekil 9. 3 No'lu Olgunun Aile Ağacı... 37
Şekil 10. 3 No'lu Olgunun Klinik Bulguları ... 37
Şekil 11. 3 No’lu Olgunun Sanger (a) ve MiSeq (b) Dizi Analizi Görüntüsü ... 38
Şekil 12. 4 No'lu Olgunun Aile Ağacı... 39
Şekil 13. 4 No'lu Olgunun Klinik Bulguları ... 40
Şekil 14. 4 No’lu Olgunun Sanger (a) ve MiSeq (b) Dizi Analizi Görüntüsü ... 40
Şekil 15. 5 No'lu Olgunun Aile Ağacı... 42
Şekil 16. 5 No'lu Olgunun Klinik Bulguları ... 42
Şekil 17. 5 No’lu Olgunun Sanger (a) ve MiSeq (b) Dizi Analizi Görüntüsü ... 43
Şekil 18. 6 No'lu Olgunun Aile Ağacı... 44
Şekil 19. 6 No'lu Olgunun Klinik Bulguları ... 45
Şekil 20. 6 No’lu Olgunun Sanger (a) ve MiSeq (b) Dizi Analizi Görüntüsü ... 45
Şekil 21. 7 No'lu Olgunun Aile Ağacı... 47
Şekil 22. 7 No'lu Olgunun Klinik Bulguları ... 47
Şekil 23. 7 No’lu Olgunun Sanger (a) ve MiSeq (b) Dizi Analizi Görüntüsü ... 48
Şekil 24. 8 No'lu Olgunun Aile Ağacı... 49
Şekil 25. 8 No'lu Olgunun Klinik Bulguları ... 50
Şekil 26. 8 No’lu Olgunun Sanger (a) ve MiSeq (b) Dizi Analizi Görüntüsü ... 50
Şekil 27. 9 No'lu Olgunun Aile Ağacı... 52
Şekil 28. 9 No'lu Olgunun Klinik Bulguları ... 52
Şekil 30. 10 No'lu Olgunun Aile Ağacı... 54
Şekil 31. 10 No'lu Olgunun Klinik Bulguları ... 55
Şekil 32. 10 No’lu Olgunun Sanger (a) ve MiSeq (b) Dizi Analizi Görüntüsü ... 55
Şekil 33. 11 No'lu Olgunun Aile Ağacı... 57
Şekil 34. 11 No'lu Olgunun Klinik Bulguları ... 57
Şekil 35. 11 No’lu Olgunun Sanger (a) ve MiSeq (b) Dizi Analizi Görüntüsü ... 58
Şekil 36. 13 No'lu Olgunun Aile Ağacı... 59
Şekil 37. 13 No'lu Olgunun Klinik Bulguları ... 60
Şekil 38. 13 No’lu Olgunun Sanger (a) ve MiSeq (b) Dizi Analizi Görüntüsü ... 60
Şekil 39. 15 No'lu Olgunun Aile Ağacı... 62
Şekil 40. 15 No'lu Olgunun Klinik Bulguları ... 62
Şekil 41. 15 No’lu Olgunun Sanger (a) ve MiSeq (b) Dizi Analizi Görüntüsü ... 63
TABLO DİZİNİ
Tablo 1. Silyopatilerde Sık Görülen Bulgular ... 5
Tablo 2. Bardet Biedl Sendromu Modifiye Tanı Kriterleri ... 7
Tablo 3. BBS Genlerinin Özellikleri ... 12
Tablo 4. PCR Primerleri ... 24
Tablo 5. Amplifikasyon İçin PCR Miksi Hazırlanışı ... 25
Tablo 6. Ampfilikasyon İçin PCR Koşulları ... 25
Tablo 7. Cycle Sequencing İçin Hazırlanan Karışım ... 28
Tablo 8. Cycle Sequencing İçin PCR Koşulları ... 28
Tablo 9. Çalışmada Saptanan Mutasyonlar ... 30
KISALTMALAR
BBS : Bardet–Biedl sendromu
LMBBS : Laurence-Moon-Bardet-Biedl sendromu IFT : İntaflagellar transport
MTOC : Mikrotübül organize edici merkez SHH : Sonic hedgehog
LMS : Laurence–Moon sendromu VKİ : Vücut kitle indeksi
BBS1 :
Bardet-Biedl syndrome 1 BBS2 :
Bardet-Biedl syndrome 2 BBS3 :
Bardet-Biedl syndrome 3 ARL6 :
ADP-ribosylation factor-like 6 BBS4 :
Bardet-Biedl syndrome 4 BBS5 :
Bardet-Biedl syndrome 5 BBS6 :
Bardet-Biedl syndrome 6 MKKS :
McKusick-Kaufman syndrome BBS7 :
Bardet-Biedl syndrome 7 BBS8 :
Bardet-Biedl syndrome 8
TTC8 :
Tetratricopeptide repeat domain 8 BBS9 :
Bardet-Biedl syndrome 9
BBS10 :
Bardet-Biedl syndrome 10 BBS11 :
Bardet-Biedl syndrome 11 TRIM32 :
Tripartite motif containing 32 BBS12 :
Bardet-Biedl syndrome 12 BBS13 :
Bardet-Biedl syndrome 13
MKS1 :
Meckel syndrome, type 1 BBS14 :
Bardet-Biedl syndrome 14 CEP290 :
Centrosomal protein 290kDa BBS15 :
Bardet-Biedl syndrome 15
WDPCP :
WD repeat containing planar cell polarity effector BBS16 :
Bardet-Biedl syndrome 16
SDCCAG8 :
Serologically defined colon cancer antigen 8 BBS17 :
Bardet-Biedl syndrome 17
LZTFL1 :
Leucine zipper transcription factor-like 1 BBS18 :
Bardet-Biedl syndrome 18
BBIP1 :
BBSome interacting protein 1
NGS : Yeni nesil dizileme (next-generation sequencing) MGPs : Magnetic Glass Particles
TD : Tagment DNA
TDE1 : Tagment DNA Enzyme ST : Stop Tagment Buffer RSB : Resuspension Buffer PMM : PCR Master Mix
NCT1 : Nextera Capture Target Buffer 1 CSO : Custom Selected Oligos
SMB : Streptavidin Magnetic Beads WS1 : Wash Solution 1
WS2 : Wash Solution 2 WS3 : Wash Solution 3 ET1 : Elute Target Buffer 1
HP3 : NaOH
ET2 : Elute Target Buffer 2 PPC : PCR Primer Cocktail
IGV : Intergrative Genomics Viewer HGMD : Human Gene Mutation Database
NCBI : National Center for Biotechnology Information NSVD : Normal spontan vajinal doğum
GÖR : Gastro özofageal reflü DM : Diyabetus mellitus ASD : Atrial septal defekt PDA : Patent duktus arteriosus
ÖZET
Bardet Biedl Sendromu (BBS) obezite, rod-kon distrofi, post aksiyal polidaktili, renal anomali, genital anomali ve öğrenme güçlüğü ile karakterize nadir görülen otozomal resesif geçişli bir silyopatidir. BBS’nin sıklığı toplumlar arası farklılık göstermektedir. KuzeyAvrupa’da 160.000’de 1 iken, akrabağa evliliğinin daha sık olduğu bazı Arap toplumlarında 13.500’de 1 görülmektedir. Türkiye’deki sıklığı ise bilinmemektedir. Bugüne kadar belirlenmiş BBS’den sorumlu olan 18 gen mevcuttur; BBS1, BBS2, BBS3 (ARL6), BBS4, BBS5, BBS6 (MKKS), BBS7, BBS8 (TTC8), BBS9, BBS10, BBS11 (TRIM32), BBS12, BBS13 (MKS1), BBS14 (NPHP6), BBS15 (WDPCP), BBS16 (SDCCAG8), BBS17 (LZTFL1), BBS18 (BBIP1). Bu çalışmada klinik olarak BBS tanısı konulan 15 olguda, hedeflenmiş yeni nesil dizi analizi ile 16 BBS geninde mutasyon dağılımı ve fenotip-genotip korelasyonu araştırılmıştır.
Çalışmaya alınan 15 olgunun 13’ünde, araştırılan genlerde hastalığa sebep olan mutasyon saptandı. BBS1 geninde, 1’i tanımlı (Y284SfsX5), 2’si yeni (IVS1-3C>G, Q338X) 3 mutasyon, BBS2 geninde 1 yeni mutasyon (G88AfsX6), BBS4 geninde 1 yeni mutasyon (IVS6-2A>G), BBS7 geninde 1’i tanımlı (R238EfsX59), 1’i yeni (L317V) 2 mutasyon, BBS9 geninde 1 yeni mutasyon (N35X), BBS10 geninde 1’i tanımlı (S311A), 3’ü yeni (K619IfsX10, I342NfsX20, T516NfsX8) 4 mutasyon saptanmıştır. Olgular klinik bulguları ve moleküler genetik özellikleri ile fenotip-genotip korelasyonu açısından değerlendirilmiştir.
Bu çalışma, BBS moleküler tanısı ve fenotip-genotip korelasyonu ile ilgili Türkiye’de ilk ve dünya literatründeki az sayıda çalışmalardan biridir. Olgulardaki moleküler bozukluğun ortaya konması, gelişebilecek komplikasyonların önlenebilmesine ve sağlıklı nesillerin yetişmesine katkı sağlayacaktır.
Anahtar Kelimeler: Bardet Biedl Sendromu, Yeni Nesil Dizi Analizi, Mutasyon, Fenotip-Genotip Korelasyonu
ABSTRACT
Bardet-Biedl Syndrome (BBS) is a rare, autosomal-recessive ciliopathy characterized by obesity, rod-cone dystrophy, postaxial polydactyly, renal abnormalities, genital abnormalities and learning difficulties. The prevalence of BBS varies between populations; from 1:160 000 in northern Europe to 1:13 500 in Arabic communities, where a higher level of consanguinity rates. The incidence is unknown in Turkey. To date, mutations in 18 different genes have been described to be responsible for BBS; BBS1, BBS2, BBS3 (ARL6), BBS4, BBS5, BBS6 (MKKS), BBS7, BBS8 (TTC8), BBS9, BBS10, BBS11 (TRIM32), BBS12, BBS13 (MKS1), BBS14 (NPHP6), BBS15 (WDPCP), BBS16 (SDCCAG8), BBS17 (LZTFL1), BBS18 (BBIP1). In this study mutations in 16 BBS genes, were investigated in 15 clinically diagnosed BBS patients via NGS technology.
In the 13 of the 15 patients studied, disease causing mutations were identified. These mutations and genes harbouring these mutations were as follows; BBS1 gene: 1 previously reported (Y284SfsX5), 2 novel (IVS1-3 C>G, Q338X) 3 mutations, BBS2 gene: 1 novel mutation (G88AfsX6), BBS4 gene: 1 novel mutation (IVS6-2 A>G), BBS7 gene: 1 previously reported (R238EfsX59), 1 novel (L317V) 2 mutations, BBS9 gene: 1 novel mutation (N35X), BBS10 gene:1 previously reported (S311A), 3 novel (K619IfsX10, I342NfsX20, T516NfsX8) 4 mutations identified. Regarding clinical features and molecular analysis results, phenotype- genotype correlation was discussed. This is the first study investigating molecular genetic etiology and phenotype- genotype correlation in patients with BBS from Turkey and one of the few studies investigating such a wide spectrum of BBS genes in BBS syndrome. Identification of molecular defects in BBS patients, prevent complications that may develop and contribute the growing of a healthy generation.
Key Words: Bardet-Biedl Syndrome, Next-Generation Sequencing, Mutation, Phenotype- Genotype Correlation
1.
Giriş ve Amaç
Silya, hücre yüzeyinde yerleşen ve hücrede anten görevi gören bir yapıdır [1]. Motil ve motil olmayan (primer) silya olmak üzere iki gruba ayrılır [1]. Motil silya hareketten sorumlu iken, primer silya sinyal iletiminden sorumludur [2]. Silya yapısında ve fonksiyonundaki hatalarından kaynaklanan hastalıklar ‘silyopatiler’ olarak isimlendirilmektedir. Silyopatilerin ortak özellikleri arasında, görme kaybı, işitme kaybı, böbrek hastalığı, kalp hastalığı, obezite ve diyabet yer almaktadır [3]. Bardet Biedl sendromu (BBS) model silyopati olarak kabul edilmektedir [4]. Silyopatiler grubunda klinik ve genetik heterojenite gösteren bugüne kadar ispatlanmış 20 hastalık ve sorumlu 95 gen yer almaktadır [5].
İlk kez 1866 yılında, Laurence ve Moon tarafından dört kardeşte retinitis pigmentosa, obezite, zeka geriliği ve spastik paraparezi bulguları olan bir aile tariflenmiştir [6]. Daha sonra, 1922 yılında Bardet ve Biedl bağımsız olarak polidaktili, obezite ve retinitis pigmentosa ile seyreden bir hastalık tariflemişlerdir [7] [8]. Önce Laurence-Moon-Bardet-Biedl sendromu (LMBBS) olarak adlandırılan hastalık, günümüzde Bardet Biedl sendromu adıyla anılmaktadır. BBS nadir bir hastalık olup sıklığı ülkeler arası farklılık göstermektedir. Avrupa ve Kuzey Amerika’da 100000-160000’de 1 görülmekte iken, bazı küçük ve akrabağa evliliğinin yüksek olduğu toplumlarda sıklık artmıştır [9]. Kuveyt Bedevileri ve Newfoundland’de sırasıyla, 13.500’de 1 ve 17.500’de 1 sıklıkta saptanmıştır [10, 11].
BBS, klinik olarak, aile içi ve aileler arası oldukça değişkenlik gösteren bir hastalıktır [12]. 1999 yılında belirlenen tanı kriterlerine göre bulgular major ve minör olmak üzere ikiye ayrılır. Major bulgular; rod-kon distrofisi, polidaktili, obezite, genital anomali, böbrek anomalisi ve öğrenme güçlüğüdür. Minör bulgular ise; konuşma sorunları, gelişme geriliği, diş sorunları, brakidaktili/sindaktili, nörolojik sorunlar, davranış sorunları, göz sorunları, poliüri/polidipsi, diabetes mellitus, anosmi, konjenital kalp hastalığıdır. Tanı için dört major bulgu ya da üç major bulgu ve iki minör bulgunun bulunması gerekmektedir [13].
BBS genetik olarak oldukça heterojenite gösteren bir hastalıktır. Bugüne kadar 18 farklı genin mutasyonları hastalıktan sorumlu tutulmuştur [14]. Mutasyonlar en sık BBS1 ve BBS10 genlerinde olup, bu iki gendeki mutasyonlar, klinik olarak tanı alan
BBS hastalarının %40’ından sorumludur [15]. BBS’ye neden olan genlerin sıklığı, etnik kökene göre değişikli göstermektedir. Örneğin BBS4 ve BBS5 mutasyonlarının çoğunlukla Orta Doğu ve Kuzey Afrika kökenli olduğu bildirilmiştir [16]. Bilinen bütün BBS genlerindeki mutasyonlar, BBS kliniği olan hastaların ancak %80’ine tanı koydurabilmektedir [17]. BBS otozomal resesif bir hastalık olmasına rağmen yaklaşık %10’unda digenik triallelik kalıtım da bildirilmiştir [18].
Yeni nesil dizileme teknolojisi xsayesinde kısa zamanda çok sayıda gen aynı anda taranabilmektedir. Bu teknoloji BBS gibi genetik heterojenitesi olan hastalıkların tanısının konması ve yeni aday gen bulunmasında büyük katkı sağlamaktadır.
Bu çalışmada klinik olarak BBS tanısı almış 15 hastada, BBS ‘ye neden olduğu belirlenmiş 16 gendeki mutasyonların, yeni nesil dizi analizi teknolojisi ile analiz ve genotipin fenotipik bulgular üzerinde etkilerinin araştırılması amaçlanmıştır.
2.
Literatür özeti
2.1 Silya yapı ve fonksiyonu
Silya hücre yüzeyinde yerleşen evrimsel olarak iyi korunmuş bir yapıdır ve hücrenin anteni olarak kabul edilmektedir [1]. İlk olarak böbrek ve tiroid dokusunda gösterilmiştir fakat günümüzde nerdeyse tüm memeli hücrelerinde olduğu bilinmektedir [19]. Motil ve motil olmayan (primer) silya olmak üzere iki gruba ayrılır [1]. Motil silya sıvıların atılımından ve hareketten sorumludur. Genellikle solunum epiteli, sperm kuyruğu, beyin ventriküllerinde yer alırlar. Motil silya 2 merkezi mikrotübül etrafında dizilmiş 9 periferik mikrotübül çiftinden oluşan ‘9+2’ aksonem yapısı gösterir. Defektinde, bronşiektazi, infertilite ve sağ-sol vücut asimetrisi ile karakterize primer silyer diskinezi hastalığı ortaya çıkmaktadır [20].
Primer silya, sinyal iletimini düzenleyen duyusal bir organel olarak görev yapar [2]. Yapısında motil silyaya benzer periferik yerleşimli 9 mikrotübül çifti yerleşir fakat merkezde mikrotübül bulunmaz [1]. Defekti primer silyopati denilen bir grup hastalıktan sorumludur. Bu grup hastalıkların ortak özellikleri arasında, görme kaybı, işitme kaybı, böbrek hastalığı, kalp hastalığı, obezite ve diyabet yer almaktadır [3]. BBS silyopatilerin prototip hastalığı olarak kabul edilmektedir [4].
Hücre yüzeyindeki silyumlar, mikrotübüllerden oluşan bir aksonem özü içermektedirler. Aksonem, mikrotübül organize edici merkez denen, sentriolden köken alan ve hücrenin apikal bölgesinde yerleşmiş bir bazal cisimden uzanmaktadır. Bazal cisim silyagenezi yönetmektedir. Silyar sitoplazmada ribozom olmadığından dolayı silya biogenezi ve fonksiyonları için gereken proteinler hücre sitoplazmasından silyum sitoplazması boyunca taşınmak zorundadır. Bu ileri-geri taşınım sistemi intaflagellar transport (IFT) olarak adlandırılmaktadır. BBS genleri tarafından kodlanan BBSome proteinleri silyagenez sürecinde yer alarak, primer silya fonksiyonunda önemli bir rol oynamaktadır [21]. BBSome proteinleri şaperon kompleks ve Rab ailesi proteinleri tarafından düzenlenmektedir [20]. Bu proteinler hep birlikte IFT’ye olanak sağlamaktadır (Şekil 1).
Silya hücre dışı sinyali moleküllerini algılıyan bir anten olarak kabul edilmektedir [1]. Sonic hedgehog (SHH), Wnt, PDGF, mTOR yolakları başta olmak üzere pek çok yolak düzgün çalışabilmesi için silyuma ihtiyaç duymaktadır [22].
Şekil 1. Primer silyanın yapısı ve intraflagellar transportta görevli genler
2.2 Silyopatiler
Silyopatiler son yıllarda bilim dünyasında oldukça sık araştırılan ve merak uyandıran konulardan biri olmuştur. Silyopatiler, silya yapısında ve fonksiyonundaki hatalardan kaynaklanan hastalıklar olarak tariflenmektedir. Bu gruptaki hastalıkların çoğu, uzun zamandır bilinen nadir hastalıklar olmasına rağmen silya disfonksiyonu ile ilişkilendirilmesi son yıllarda olmuştur [1]. İlk kez 2003 yılında Ansley ve ark. tarafından BBS8 genini tarifledikleri makalede ‘silyopati’ adından bahsedilmiştir[23]. Silyum hastalıklarının kliniği çok geniş olmakla birlikte genellikle, retinopati, kistik
böbrek hastalığı, zeka geriliği, polidaktili, santral sinir sistemi anomalileri, karaciğer hastalığı, obezite, anosmi, kalp hastalığı, hipogonadizm, situs inversus ve sağırlık görülmektedir. Fizyolojisi henüz tam olarak anlaşılamamış olan bu grupta farklı dokulardaki silya fonksiyonlarının kalitatif ve kantitatif değişikliğinin kliniği etkilediği düşünülmektedir. Aynı gendeki mutasyonların farklı sendromlara yol açması da bu teoriyi desteklemektedir [24, 25].
Silyopatiler, silya tipine göre (motil,primer) yada fenotipik bulgularına göre (iskelet tutulumu, böbrek hastalığı vb.) sınıflandırılabilir[1]. Primer silyopatiler pek çok ortak klinik bulguya sahiptir (Tablo 1). Silyopati olduğu kanıtlanmış hastalıklar gün geçtikçe artmaktadır. Son kesinleşmiş bilgilere göre 20 silyopati ve sebep olan 95 gen mevcuttur [5]. Tablo 1. Silyopatilerde Sık Görülen Bulgular
Hastalık BBS OFD1 Meckel Joubert Jeune
Retinitis pigmentosa + + + +
Kistik böbrek hastalığı + + + + +
Polidaktili + + + + +
Situs inversus + + +
Zeka geriliği + + + +
Karaciğer hastalığı + + + +
İskelet bozukluğu + + + +
Arka beyin anomalisi + +
BBS: Bardet Biedl sendromu, OFD 1: Oro-fasio-dijital sendrom tip 1
2.3 Bardet Biedl sendromunun tarihçesi
İlk kez 1866 yılında, John Z. Laurence (1829-1870) ve Robert Moon (1844-1914) isimli iki göz hastalıkları uzmanı tarafından yayınlanan olgu sunumunda dört kardeşte
retinitis pigmentosa, obezite, zeka geriliği ve spastik paraparezi bulguları olan bir aile tariflenmiştir [6]. 1922 yılında ise George Bardet ve Artur Biedl bağımsız olarak polidaktili, obezite ve retinitis pigmentosa bulgularıyla giden bir hastalık tariflemişlerdir [7] [8]. Birkaç yıl sonra Solis-Cohen ve Weiss, Laurence ve Moon ile Bardet ve Biedl’ın yayınladığı olguların aynı antite olduğunu fark etmişler ve bu sendrom ‘Laurence-Moon-Bardet-Biedl sendromu’ (LMBBS) olarak isimlendirmişlerdi. Son zamanlarda sendrom iki ayrı gruba ayrılmıştır: Laurence–Moon sendromu (LMS) ve Bardet–Biedl sendromu. Laurence Moon sendromu obezite ve polidaktili olmaksızın retinis pigmentosa, mental retardasyon, hipogenitalizm ve spastic paraparezi ile karakterizedir. Bardet Biedl sendromunun ana bulguları; retinis pigmentosa, postaksiyel polidaktili, obezite, mental retardasyon, hipogenitalizm olup paraparezi bu grupta yer almamaktadır. Ortak fenotipik bulguların varlığı nedeniyle allelik hastalıklar olabileceği düşünülmektedir [13]. Yayınlamış olguların çoğunluğu da göz önüne alınarak Bardet Biedl sendromu rutin olarak kullanılan isimlendirme haline gelmiştir.
2.4 Prevalans
BBS nadir bir genetik hastalık olup, sıklığı ülkeler arası farklılık göstermektedir. Avrupa ve Kuzey Amerika’da sıklık 100.000’de 1 (Kuzey Amerika) ile 160.000’de 1 (İsviçre) arasında görülmektedir [26].
Akrabağa evliliğinin sık olduğu Kuveyt Bedevileri’nde 13.500’de 1 sıklıkta saptanmıştır [10]. İzole bir ada olan Newfoundland’de ise 17.500’de 1 sıklıkta görülmektedir [11]. Prevelansın toplumlar arasında farklılık göstermesi kurucu etkisi ile açıklanabilmektedir.
2.5 Bardet Biedl sendromunun klinik özellikleri
BBS klinik olarak oldukça değişkenlik gösteren bir hastalık olup hayatın ilk on yılında yavaş yavaş ortaya çıkmaktadır. Genellikle doğumda saptanan polidaktili ile ilk olarak BBS tanısı akla gelmektedir. Polidaktili sık saptanan bir bulgudur ve ek parmak anomalileri eşlik edebilmektedir. Retinitis pigmentosa ve obezite gibi bulgular ise erken çocukluk ya da
ergenlik döneminde ortaya çıkabilmektedir. Ortalama tanı yaşı farklı yayınlarda 9 ile 15 yaş arası bildirilmektedir [13, 27]. Ailelerin çocuklarında bir sorun olduğunu ilk fark etme zamanı ise 3 yaş civarı olarak bildirilmiştir [13]. Pek çok semptomu barındıran bir sendrom olması nedeniyle, tanı için objektif kriterler olması son derece önemlidir. Beals ve ark. tarafından 1999 yılında İngiltere’de 109 BBS olgusu ve ailelerinde yapılan çalışma sonucu tanı kriterleri yenilenmiştir [13]. Major bulgular; rod-kon distrofisi, polidaktili, obezite, genital anomali, böbrek anomalisi ve öğrenme güçlüğüdür. Minör bulgular arasında ise; konuşma sorunları, gelişme geriliği, diş sorunları, brakidaktili/sindaktili, nörolojik sorunlar, davranış sorunları, göz sorunları, poliüri/polidipsi, diabetes mellitus, anosmi, konjenital kalp hastalığı sayılabilir (Tablo 2). Tanı için dört major bulgu ya da üç major bulgu ve iki minör bulgunun bulunması gerekmektedir.
Tablo 2. Bardet Biedl Sendromu Modifiye Tanı Kriterleri
Major kriterler Minör kriterler
Rod-kon distrofisi Konuşma sorunları
Polidaktili Gelişme geriliği
Obezite Diş sorunları
Genital anomali Brakidaktili/sindaktili
Böbrek anomalisi Nörolojik sorunlar
Öğrenme güçlüğü Davranış sorunları
Göz sorunları Poliüri/polidipsi Diabetes mellitus Anosmi
Konjenital kalp hastalığı Astım
2.5.1Major bulgular
2.5.1.1 Obezite
Obezite BBS olgularının %72 ile %92’sinde saptanmaktadır [15]. Doğum kiloları üst sınıra yakın olmasa da normal sınırlardadır [28]. Obezite çocukluk çağlarında başlayıp yaşla birlikte artar. Ortalama vücut kitle indeksi (VKİ) kadınlarda 31.5 mg/m2, erkeklerde ise 36.6 mg/m2 olarak saptanmıştır [29]. Yapılan çalışmada puberte sonrası hastaların %20’si kilolu (VKİ>25 kg/m2), %36’sı obez (VKİ>30 kg/m2) ve %16’sı morbid obez (VKİ>40 kg/m2) bulunmuştur [13]. Çocukluk çağında yaygın obezite görülürken yetişkinlikte trunkal obezite ön plandadır [15].
2.5.1.2 Rod-kon distrofisi
Bu grup bozukluklar kon ve rod fotoreseptörlerinde bozukluk ile birliktedir ve genellikle çocukluk çağında başlamaktadır. İlk olarak rod fotoreseptörlerinin ve daha sonra kon fotoreseptörlerinin kaybı ile karakterizedir [30]. Erken maküler tutulumla giden atipik retinitis pigmentosa ortaya çıkmaktadır ve erken dönemlerden itibaren ciddi görme kayıpları izlenmektedir [1]. Gece görüş zorluğu, fotofobi, renkli görmede bozulma ve görme alanında daralma görülebilmektedir [30].
BBS tanısı için en sık kullanılan klinik bulgu rod-kon distrofisidir. Hastaların %90’ından fazlasında saptanmaktadır [15]. Tipik bulgularını taşıyabildiği gibi, tersten önce kon sonra rod fotoreseptörlerinin kaybı da görülebilmektedir. Yapılan bir çalışmada ailelerin ilk kez gece körlüğünü fark ettiği yaş ortalama 8.5 iken hastaların kör olarak bildirimi ortalama 15.5 yaş olarak saptanmıştır [13]. BBS’de kliniğin hızlı ilerlediği ve morbiditenin en önemli sebeplerinden birini oluşturduğu bilinmektedir.
2.5.1.3 Polidaktili
Postaksiyel polidaktili fazla parmağın 5. parmağın distalinde yerleşmesi olarak tanımlanmaktadır. Sporadik olarak görülebilen bir malformasyon olabilmesine karşın BBS’nin doğumda saptanabilen tek bulgusu da olabilmektedir. Cerrahi olarak çıkarılabilmesi ve genellikle ekstremite fonksiyonlarını bozmaması nedeniyle en az morbidite sebebi olan bulgu olarak sayılabilir. Sıklığı %63 ile %81 arasında bildirilmiştir [15]. Tek bir ekstremiteyi tutabildiği gibi dört ekstremite de görülebilmektedir. Yapılan çalışmalar sonucu sadece üst ekstermitede %8, sadece alt ekstremitede %21 ve dört ekstremitede %21 olarak görüldüğü saptanmıştır [13].
Polidaktiliye diğer parmak anomalileri de eşlik edebilmektedir. Kısa, kalın, güdük parmaklar hastaların yaklaşık yarısında görülmektedir [13]. Ayrıca sindaktili de görülebilmektedir.
2.5.1.4 Genital anomali
Deveault ve ark. tarafından yapılan multi-etnik kohort çalışmasına göre genital anomali görülme sıklığı %59 olarak saptanmıştır [31]. Erkeklerde hipogonadizme bağlı puberte gecikmesi ve hipogenitalizm görülmektedir. Testis volümünde azalma, küçük penis ve kriptorşidizm bildirilmiştir [13]. Kadınlarda ise yapısal genital anomaliler ön planda saptanmıştır. Bunlar arasında hipoplastik uterus, tuba uterina ve overler, vajinal atrezi, hidro/hidrometrokolpos, persistan ürogenital sinüs, vezikovajinal fistül sayılabilmektedir [32, 33]. Erkekler daima infertil iken, kadınlarda azalmış da olsa fertilite bildirilmiştir [13].
2.5.1.5 Böbrek anomalisi
Böbrek anomalileri morbidite ve mortalitenin ön önemli sebebini oluşturmaktadır [34]. Böbrek anomalilerinin görülme sıklığı yaklaşık %50 olarak saptanmıştır [15].
Klinik varyasyon göstermekle birlikte genellikle asemptomatiktir. Klasik olarak kistik tübüler hastalık ve anatomik malformasyonlar saptanmakta, anemi, poliüri ve polidipsi kliniği ile bulgu vermektedir [28]. Daha nadir olarak, fokal segmental glomerüloskleroz ve glomerül bazal membran ayılması ile karakterize glomerüler hastalık da bildirilmiştir [35]. Hastaların çoğunda yapısal anomali olmamasına ve böbrek fonksiyonları normal olmasına rağmen idrar konsantrasyon defektleri gözlenmektedir [36]. Çocukluk çağında olguların üçte birinde ilk saptanan bulgu olan poliüri-polidipsi, vazopressin dirençli üriner konsantrasyon defektine bağlı olarak görülür [28, 37, 38].
2.5.1.6 Öğrenme güçlüğü
BBS hastalarında gelişme geriliği ve zihinsel yetersizliksizlik sık görülmektedir. Beales ve ark.’nın 109 hastalık kohortunda, hastaların %62’sinde zihinsel yetersizliksizlik olduğu ve bunların yarısının özel eğitime gittiği saptanmıştır [13]. Yapılan pek çok çalışma sonucu hastaların çoğunda belirgin öğrenme güçlüğü saptanmasına karşın IQ testinde sadece ufak bir grupta ciddi gerilik saptanmıştır [13, 29, 39]. Kısa süreli belleğin zayıf olduğu fakat ayrıntılarla ilgili uzun süreli belleğin çok iyi olduğu bildirilmiştir. Davranış sorunları, özellikle obsesif kompulsif davranışlarla sık karşılaşılmakta ve nadiren otizm ya da psikoz gibi daha ağır sorunlar görülebilmektedir [13].
2.5.2Minör bulgular
BBS’de pek çok farklı ve daha nadir görülen minör bulgu bildirilmiştir. Minör bulgular tanı koymada oldukça faydalı olmaktadır. Sık görülenler; konuşma sorunları, gelişme geriliği, diş sorunları, parmak anomalileri, nörolojik sorunlar, davranış sorunları, poliüri/polidipsi, diabetes mellitus, anosmi, konjenital kalp hastalığı ve astımdır.
2.6 Bardet Biedl sendromunda genetik heterojenite
BBS genetik heterojenite gösteren bir hastalıktır. İlk kez akrabalığın sık olduğu Arap Bedevileri’nde yapılan bir bağlantı analizi çalışması ile BBS geni tanımlanmıştır[40]. Bugüne kadar bildirilen on sekiz BBS lokusu mevcuttur. Bunlar: BBS1 (11q13.2) [41], BBS2 (16q12.2) [40], BBS3 (3p11.2) [42], BBS4 (15q24.1) [43], BBS5 (2q31.1) [44], BBS6 (20p12.2) [44], BBS7 (4q27) [45], BBS8 (14q31.3) [23], BBS9 (7p14.3) [46], BBS10 (12q21.2) [47], BBS11 (9q33.1) [48], BBS12 (4q27) [17], BBS13 (17q22) [25], BBS14 (12q21.32) [25], BBS15 (2p15) [49], BBS16 (1q43) [50], BBS17’dir (3p21.31) [51], BBS18 (10q25.2) [14] (Tablo 3).
Bildirilen on sekiz BBS geni olmasına rağmen, patojenik mutasyonların çoğu BBS1 ve BBS10’da saptanmıştır. Bu iki gen klinik olarak tanı alan BBS hastalarının %40’ını oluşturmaktadır [15]. En sık rastlanan mutasyonlar, BBS1’de M309R ve BBS10’da C91LfsX5 mutasyonlarıdır [15]. Bazı genlerin etnik sıklık dağılımı diğerlerinden daha belirgindir. Örneğin BBS4 ve BBS5 mutasyonlarının çoğunlukla Orta Doğu ve Kuzey Afrika kökenli olduğu bildirilmiştir [52, 53]. BBS3 ve BBS8 genleri gibi bazı BBS genlerinin mutasyonları çok nadir olup, sendromun %5’inden daha azından sorumludur [23, 54]. BBS11 ve BBS14 genleri ise sadece birer ailede tariflenmiştir [25, 48]. Klinik olarak tanı konan BBS hastalarının yaklaşık %20’sinde bilinen BBS genlerinde mutasyon saptanmamıştır [17]. Bu durum, henüz tanımlanmamış yeni genlerin varlığını düşündürmektedir.
BBS otozomal resesif bir hastalık olmasına rağmen kliniğin ortaya çıkabilmesi için ikinci bir lokusta üçüncü bir mutasyon olması gerektiği durumlar bildirilmiştir (triallelizm) [18]. Örneğin, Katsanis ve ark.’nın çalışmasında BBS2’de Q59X ve Y24X mutasyonlarını birleşik heterozigot olarak taşıyan birey fenotipik olarak normalken, BBS kliniği gösteren kardeşinde bu iki mutasyonun yanı sıra BBS4’te Q147X mutasyonu taşıdığı saptanmıştır [55]. Yapılan pek çok çalışmada ise triallelizm hiç saptanmamıştır [56, 57] [58]. BBS’da triallelizm sıklığının %10’un altında olduğu düşünülmekte bu nedenle ailede moleküler olarak kanıtlanmış triallelizm olgusu yoksa otozomal resesif hastalık risk değerlendirmesine göre genetik danışma verilmesi ve nadiren farklılık olabileceğinin aileye bildirilmesi önerilmektedir [59].
Tablo 3. BBS Genlerinin Özellikleri
Gen Lokus Sıklık Fonksiyon
BBS1 11q13.2 %23 BBSome proteini BBS2 16q12.2 %8 BBSome proteini BBS3/ARL6 3p11.2 %0.4 GTPaz BBS4 15q24.1 %2 BBSome proteini BBS5 2q31.1 %0.4 BBSome proteini BBS6/MKKS 20p12.2 %6 Şaperon kompleks BBS7 4q27 %2 BBSome proteini BBS8 14q31.3 %1 BBSome proteini BBS9 7p14.3 %6 BBSome proteini BBS10 12q21.2 %20 Şaperon kompleks
BBS11/TRIM32 9q33.1 %0.1 E3 ubiquitin ligaz
BBS12 4q27 %5 Şaperon kompleks
BBS13/MKS1 17q22 %4.5 Sentriol migrasyonu
BBS14/CEP290 12q21.32 %1 Gate keeper
BBS15/WDPCP 2p15 %1 Silyogenez
BBS16/SDCCAG8 1q43 %1 OFD1 ile etkileşim
BBS17/LZTFL1 3p21.31 <%1 Negatif regülatör
2.7 Bardet Biedl sendromunda genotip-fenotip korelasyonu
BBS genotip-fenotip korelasyonu zayıf olan hastalıklardan birisidir. Yapılan bir çalışmada BBS1 geninde saptanan en sık mutasyon olan M390R hafif feanotiple ilişkilendirilmiştir [58]. Bazı spesifik göz bulgularını ve ciddi parmak anomalilerini BBS2, BBS3 ve BBS4 genleri ile ilişkilendiren yayınlar mevcuttur [60, 61]. Geniş hasta popülasyonuyla yapılan çalışmalar bu bulguları desteklememektedir. Aile içi ve aileleler arası çalışmalarda geniş fenotipik değişkenlik saptanmıştır. Bu durum BBS proteinlerinin ortak hücresel süreçleri etkilediği ve bu sebeple genotipin klinik olarak ayırt edilemediği hipotezini desteklemektedir [62].
1.6 Yeni nesil dizileme
1977 yılında Sanger tarafından bulunan dideoksi nükleotid ile zincir sonlanması yöntemi DNA sekanlamasında dönüm noktası oluşturmuştur[63]. Son 20 yıldır aynı yöntemden geliştirilen otomatize Sanger sekanslama metodu kullanılmaktadır. Bu süreçte daha uzun DNA fragmanlarını eş zamanlı okuma özellikleri geliştirilmiştir. 1990 yılında insan genomunun tamamını göstermek amacıyla başlatılan İnsan Genom Projesi’nin asıl teklonojisi de otomatize Sanger sekanslama yöntemidir. Bu projenin ilk sonuçlarını alması yaklaşık 10 yıl sürmüş, tamamlanması için ise 3 yıl daha gerekmiştir. Sonraki yıllar içinde HapMap Projesi (International HapMap Project [2006]). ve 1000 Genom Projesi’nin (1000 Genomes [2008]) de içinde olduğu benzer çalışmalar yapılmıştır. Yoğun sekanslama gerektiren bu projeler daha ekonomik, kaliteli ve hızlı yeni sekanlama teknolojilerinin geliştirilmesine neden olmuştur [64, 65]. Yeni nesil dizileme olarak isimlendirilen teknoloji, çok yüksek bir hızda ve düşük maliyette sekanslamayı olanaklı hale getirmiştir.
Yeni nesil dizileme sistemi ile dizileme milyonlarca küçük DNA parçacıkları ve adaptör dizilerinin, pikolitre hacimlerde, büyük çaplı paralel dizileme (massive parelel sequencing) ile dizilenmesi prensibine dayanmaktadır [66]. Yeni nesil dizileme teknolojisi hayal bile edilemeyecek kadar çok bilgi ve yeni yaklaşımlar sağlamaktadır.
Fakat bu kadar çok bilginin depolanması, analizi ve değerlendirmesinde büyük güçlükler vardır. Yeni nesil dizileme teknolojisinin başarılı bir şekilde kullanılması için gelişmiş biyoinformatik analiz araçlarına ihtiyaç duyulmaktadır [67].
Yeni nesil dizileme özellikle genetik heterojenitesi olan Mendelyan hastalıkların genetik sebebinin belirlenmesinde devrim yaratmıştır [68]. Klinik ile ilişkili genleri içeren ekzom panelleri tanı koyma oranını yükseltmiş ve gereken zamanı düşürmüştür. Tüm ekzom yaklaşımı ise hem bilinen genlerdeki mutasyonları ortaya koymakta hem de yeni aday gen bulmayı kolaylaştırmayı başarmaktadır.
3.
GEREÇ ve YÖNTEM
3.1 Olgu Seçimi
Ocak 2012 - Nisan 2014 tarihleri arasında Ege Üniversitesi Tıp Fakültesi Hastanesi Çocuk Sağlığı ve Hastalıkları Anabilim Dalı, Çocuk Genetik Bilim Dalı’nda ve Tıbbi Genetik Anabilim Dalı’na klinik bulgular, dismorfik bulgular, radyoloji ve laboratuar sonuçları ile BBS ön tanısı alan 15 hasta çalışmaya alındı.
Hastalar muayene edilerek ve medikal kayıtları taranarak elde edilen bilgiler, standard bir forma kaydedildi. Bu formlar hastaların dismorfik bulguları, fizik muayene bulguları, laboratuar bulguları, özgeçmiş bilgileri, soygeçmiş bilgileri ve aile ağacını içermekteydi.
Ege Üniversitesi Bilimsel Araştırma Projeleri Genel Müdürlüğü tarafından desteklenen çalışmanın Ege Üniversitesi Tıp Fakültesi Araştırma Etik Kurulu onayı (2014-TIP-012 nolu) alındı. Fotoğraflarının ve klinik bilgilerinin yayınlanmasına izin verdiklerini belirten gönüllü olur formu imzalatıldıktan sonra hasta grubundan ve ebeveynlerinden 2 ml EDTA’lı tüpe kan örnekleri alındı. Örnekler, örnek toplama işlemi tamamlanıncaya kadar -20°C derecede saklandı. Toplanan örneklerden BBS’dan sorumlu olan BBS1, BBS2, BBS3 (ARL6), BBS4, BBS5, BBS6 (MKKS), BBS7, BBS8 (TTC8), BBS9, BBS10, BBS11 (TRIM32), BBS12, BBS13 (MKS1), BBS14 (CEP290), BBS15 (WDPCP), BBS16 (SDCCAG8) genlerinde mutasyon taraması yapıldı.
3.2 Olgularda Moleküler Genetik Çalışma
Çalışmaya dahil edilen olgularda 16 gen NGS ile dizi analizi yöntemi ile incelendi. Bu yöntem için öncelikle olguların DNA’ları İllumina TruSight Exome hedef zenginleştirme kiti ile çoğaltıldı. Son ürünler yeni nesil dizileme cihazına yüklenerek analiz edildi. Çıkan sonuçlar Sanger dizi analizi yöntemi ile doğrulandı. Ebeveyn DNA’ları da Sanger dizi analizi yöntemi ile taşıyıcılığı göstermek için analiz edildi.
3.2.1Örneklerin Toplanması ve DNA İzolasyonu
Olgulardan EDTA’lı tüpe 2cc venöz kan örneği alındı. Kan lenfosit hücrelerinden protokole uygun olarak, aşağıda yazılı adımlar izlenerek QIAcube AllPrep DNA/RNA FFPE kit (Product No:80234; QIAGEN, Germany) kiti kullanılarak QIAcube Robotik DNA izolasyon cihazı ile DNA izolasyonu yapıldı.
1. 200 µl kan örnek kartuşu içine aktarıldı.
2. Örnek QIAcube Robotik DNA İzolasyon Cihazındaki örnek kartuşu bölgesine yerleştirildi.
3. Kaç adet DNA izolasyonu yapılacağına bağlı olarak diğer süspansiyonlardan ne kadar ekleneceği sistem tarafından belirlenerek tavsiye edilen miktarda Magnetic Glass Particles (MGPs) Süspansiyonu, Proteinaz K, Elution Buffer, Wash Buffer 1 ve Wash Buffer 2 solüsyonları programda gösterildiği gibi uygun reagent kartuşlarına yüklenerek uygun yerlere yerleştirildi.
4. Örnekler ve kullanılan diğer kimyasallar yerleştirildikten sonra sistemin kullanacağı steril pipet uçları programda belirtilen yerine yerleştirildi.
5. Program çalıştırıldı. Tüm işlemler cihaz tarafından otomatik olarak gerçekleştirildi.
6. Elde edilen 100 µl DNA’lar DNA saklama kutularına konularak -20° C ’ de saklandı.
İzolasyon sonrası her örnekten 100 mikrolitre, ortalama konsantrasyonları 30 ng/ µl, saflıkları (A260/280 değeri) ortalama 1.8’in üzerinde olan DNA elde edildi.
3.2.2Araştırılan DNA Bölgelerinin Çoğaltılması
Elde edilen DNA’lardan çalışılan gen bölgeleri TruSight Exome Hedef Zenginleştirme Kiti (Illumina, San Diego, CA) ile çoğaltıldı. Protokole uygun olarak, aşağıda yazılı adımlar izlendi.
1- DNA tegmentasyonu
1. Her örnek için 2.5 ng/µl konsantrasyondaki DNA’dan 20 µl DNA 0.2’lik eppendorf tüpe alındı.
2. Üzerine 25 µl Tagment DNA (TD) Buffer eklendi.
3. Üzerine 5 µl Tagment DNA Enzyme (TDE1) eklenip 10 kez pipetej yapıldı. 4. 280 xg hızda 200 C’de 1 dakika santrifüj edildi.
5. Daha sonra PCR tüpleri PCR aletine yerleştirilerek aşağıda verilen koşullarda inkübasyon sağlandı.
550 C’de 5 dakika 100 C’de ∞
2- Tegmente DNA’nın temizlenmesi
6. İnkübasyon işleminden sonra örneklere 15 µl Stop Tagment Buffer (ST) eklenip 10 kez pipetaj yapıldı.
7. Oda ısısında 5 dakika inkübe edildi.
8. 280 xg hızda 200 C’de 1 dakika santrifüj edildi.
9. Tüplerin üzerine 52 µl AMPure XP Beads eklenip 10 kez pipetaj yapıldı. 10. Oda ısısında 10 dakika inkübe edildi.
11. 280 xg hızda 200 C’de 1 dakika santrifüj edildi. 12. Manyetik standde 2 dakika bekletildi.
13. Supernatant kısmı atıldı.
14. Manyetik standden kaldırmadan 200 µl taze hazırlanmış %80’lik etanol eklendi ve 30 saniye bekletildi.
15. Supernatant kısmı atıldı.
16. 14 ve 15. basamaklar tekrarlandı.
17. Manyetik standden kaldırmadan oda ısısında 10 dakika kurumaya bırakıldı. 18. Manyetik standden kaldırılıp 22.5 µl Resuspension Buffer (RSB) eklendi ve 10 kez pipetaj yapıldı.
19. Manyetik standde 2 dakika bekletildi. 20. 20 µl supernatant yeni bir tüpe alındı.
3- PCR amplifikasyonu
21. 19.basamakta elde edilen 20 µl ürüne aşağıdaki oranlarda hazırlanan mix katıldı. 20 µl 10x PCR Master Mix (PMM)
5 µl İndex1 5 µl İndex 2
22. 280 xg hızda 200 C’de 1 dakika santrifüj edildi.
23. Daha sonra PCR tüpleri PCR aletine yerleştirilerek aşağıda verilen koşullarda inkübasyon sağlandı. 720 C’de 3 dakika 980 C’de 30 saniye 980 C’de 10 saniye 600 C’de 30 saniye 10 döngü 720 C’de 30 saniye 720 C’de 5 dakika 100 C’de ∞ 4- PCR temizleme
24. Ürün PCR aletinden çıkarılarak 280 xg hızda 200 C’de 1 dakika santrifüj edildi. 25. Üzerine 45 µl AMPure XP Bead eklendi ve 10 kez pipetaj yapıldı.
26. Oda ısısında 10 dakika inkübe edildi. 27. Manyetik standda 2 dakika bekletildi. 28. Supernatant kısmı atıldı.
29. Manyetik standdan kaldırmadan 200 µl taze hazırlanmış %80’lik etanol eklendi. 30. Supernatant kısmı atıldı.
31. 28 ve 29. basamaklar tekrarlandı.
32. Manyetik standdan kaldırmadan oda ısısında 15 dakika kurumaya bırakıldı. 33. Manyetik standdan kaldırılıp 40 µl Resuspension Buffer (RSB) eklendi ve 10 kez pipetaj yapıldı.
34. Manyetik standda 2 dakika bekletildi. 35. 38 µl supernatant yeni bir tüpe alındı.
36. Bütün ürünlerin DNA konsantrasyonu Qubit DNA konsantrasyon ölçme cihazı ile ölçüldü. Her bir örnekten en az 500 ng DNA içerecek ve toplam volüm 40 µl olacak şekilde birleştirildi.
5- İlk hibridizasyon
37. 36. basamakta elde edilen 40 µl ürüne aşağıdaki oranlarda hazırlanan mix katıldı.
50 µl Nextera Capture Target Buffer 1 (NCT1)
10 µl TruSight Content Set Custom Selected Oligos (CSO) 38. 280 xg hızda 200 C’de 1 dakika santrifüj edildi.
39. Daha sonra PCR tüpleri PCR aletine yerleştirilerek aşağıda verilen koşullarda inkübasyon sağlandı.
950 C’de 10 dakika 18 siklus
930 C’de 1 dakika, her siklus 20 C düşerek 580 C’de 16-20 saat
6- İlk yıkama
40. Ürün PCR aletinden çıkarılarak 280 xg hızda 200 C’de 1 dakika santrifüj edildi. 41. 250 µl Streptavidin Magnetic Beads (SMB) eklendi ve 20 kez pipetaj yapıldı. 42. Oda ısısında 30 dakika inkübe edildi.
43. 280 xg hızda 200 C’de 1 dakika santrifüj edildi. 44. Manyetik standda 2 dakika bekletildi.
45. Supernatant kısmı atıldı ve manyetik standdan kaldırıldı.
46. 200 µl Wash Solution 1 (WS1) eklendi ve 20 kez pipetaj yapıldı. 47. Manyetik standde 2 dakika bekletildi.
48. Supernatant kısmı atıldı ve manyetik standdan kaldırıldı.
49. 200 µl Wash Solution 2 (WS2) eklendi ve 20 kez pipetaj yapıldı. 50. Manyetik standda 2 dakika bekletildi.
51. Supernatant kısmı atıldı ve manyetik standdan kaldırıldı. 52. Tekrar 200 µl WS2 eklendi ve 20 kez pipetaj yapıldı.
53. PCR tüpü PCR aletine yerleştirilerek 420 C’de 30 dakika inkübasyonu sağlandı. 54. Ürün PCR aletinden çıkarılarak hızlıca manyetik stande alınıp 2 dakika bekletildi.
55. Supernatant kısmı atıldı ve manyetik standdan kaldırıldı. 56. 52-55. basamaklar tekrarlandı.
57. 200 µl Wash Solution 3 (WS3) eklendi ve 20 kez pipetaj yapıldı. 58. Manyetik standda 2 dakika bekletildi.
59. Supernatant kısmı atıldı ve manyetik standdan kaldırıldı. 60. 57-59. basamaklar tekrarlandı.
61. Ayrı bir PCR tüpünde aşağıdaki oranlarda mix hazırlandı. 28.5 µl Elute Target Buffer 1 (ET1)
1.5 µl 2N NaOH (HP3) 30 µl total volüm
62. 60. basamakta elde edilen ürüne 23 µl hazırlanan miks eklendi ve 20 kez pipetaj yapıldı.
63. Oda ısısında 5 dakika inkübe edildi.
64. 280 xg hızda 200 C’de 1 dakika santrifüj edildi. 65. Manyetik standda 2 dakika bekletildi.
66. 21 µl supernatant yeni bir tüpe alındı.
67. Üzerine 4 µl Elute Target Buffer 2 (ET2) eklendi ve 10 kez pipetaj yapύldύ.
7- İkinci hibridizasyon
68. 67. basamakta elde edilen 25 µl ürüne aşağıdaki oranlarda hazırlanan miks katıldı.
50 µl NCT1 10 µl CSO 15 µl ddH2O
69. 280 xg hızda 200 C’de 1 dakika santrifüj edildi.
70. Daha sonra PCR tüpleri PCR aletine yerleştirilerek aşağıda verilen koşullarda inkübasyon sağlandı.
930 C’de 1 dakika, her döngü 20 C düşerek 18 döngü 580 C’de 16-20 saat
8- İkinci yıkama
71. Ürün PCR aletinden çıkarılarak 280 xg hızda 200 C’de 1 dakika santrifüj edildi. 72. 250 µl SMB eklendi ve 20 kez pipetaj yapıldı.
73. Oda ısısında 30 dakika inkübe edildi.
74. 280 xg hızda 200 C’de 1 dakika santrifüj edildi. 75. Manyetik standda 2 dakika bekletildi.
76. Supernatant kısmı atıldı ve manyetik standdan kaldırıldı. 77. 200 µl WS1 eklendi ve 20 kez pipetaj yapıldı.
78. Manyetik standda 2 dakika bekletildi.
79. Supernatant kısmı atıldı ve manyetik standdan kaldırıldı. 80. 200 µl WS2 eklendi ve 20 kez pipetaj yapıldı.
81. Manyetik standda 2 dakika bekletildi.
82. Supernatant kısmı atıldı ve manyetik standdan kaldırıldı. 83. Tekrar 200 µl WS2 eklendi ve 20 kez pipetaj yapıldı.
84. PCR tüpü PCR aletine yerleştirilerek 420 C’de 30 dakika inkübasyonu sağlandı. 85. Ürün PCR aletinden çıkarılarak hızlıca manyetik standa alınıp 2 dakika bekletildi.
86. Supernatant kısmı atıldı ve manyetik standdan kaldırıldı. 87. 83-86. basamaklar tekrarlandı.
88. 200 µl WS3 eklendi ve 20 kez pipetaj yapıldı. 89. Manyetik standda 2 dakika bekletildi.
90. Supernatant kısmı atıldı ve manyetik standdan kaldırıldı. 91. 88-90. basamaklar tekrarlandı.
92. Ayrı bir PCR tüpünde aşağıdaki oranlarda miks hazırlandı. 28.5 µl ET1
1.5 µl 2N HP3 30 µl total volüm
93. 91. basamakta elde edilen ürüne 23 µl hazırlanan miks eklendi ve 20 kez pipetaj yapıldı.
94. Oda ısısında 5 dakika inkübe edildi.
95. 280 xg hızda 200 C’de 1 dakika santrifüj edildi. 96. Manyetik standda 2 dakika bekletildi.
97. 21 µl supernatant yeni bir tüpe alındı.
98. Üzerine 4 µl ET2 eklendi ve 10 kez pipetaj yapıldı.
9- PCR amplifikasyonu
99. 98. basamakta elde edilen 25 µl üründen 20 µl alınarak aşağıdaki oranlarda hazırlanan miks eklendi.
25 µl PMM
5 µl PPC (PCR Primer Cocktail)
100. 280 xg hızda 200 C’de 1 dakika santrifüj edildi.
101. Daha sonra PCR tüpleri PCR aletine yerleştirilerek aşağıda verilen koşullarda inkübasyon sağlandı. 980 C’de 30 saniye 980 C’de 10 saniye 600 C’de 30 saniye 12 döngü 720 C’de 30 saniye 720 C’de 5 dakika 100 C’de ∞
102. Ürün PCR aletinden çıkarılarak üzerine 90 µl AMPure XP Bead eklendi ve 10 kez pipetaj yapıldı.
103. Oda ısısında 15 dakika inkübe edildi. 104. Manyetik standda 5 dakika bekletildi. 105. Supernatant kısmı atıldı.
106. Manyetik standdan kaldırmadan 200 µl taze hazırlanmış %80’lik etanol eklendi ve 30 saniye bekletildi.
107. Supernatant kısmı atıldı.
108. 106 ve 107. basamaklar tekrarlandı.
109. Manyetik standdan kaldırmadan oda ısısında 15 dakika kurumaya bırakıldı. 110. Manyetik standdan kaldırılıp 30 µl Resuspension Buffer (RSB) eklendi ve 10 kez pipetaj yapıldı.
111. Oda ısısında 2 dakika inkübe edildi. 112. Manyetik standda 5 dakika bekletildi. 113. 28 µl supernatant yeni bir tüpe alındı.
114. Bütün ürünlerin DNA konsantrasyonu Qubit DNA konsantrasyon ölçme cihazı ile ölçüldü.
115. Son üründen 15 ng DNA içerecek şekilde uygun koşullarda MiSeq v2 300 döngülük kartuşa yüklendi.
116. Kartuş MiSeq cihazına yerleştirilip uygun program seçilerek çalıştırıldı.
Örnekler IGV programında, insan genomu ‘hg19’ referans alınarak değerlendirildi. Sonuçlar programında değerlendirildi. Saptanan değişiklikler Human Gene Mutation Database (HGMD), Ensembl ve National Center for Biotechnology Information (NCBI) veritabanları kullanılarak değerlendirildi. İlk kez saptanan varyantlar Mutation Taster ve PolyPhen-2 modelleme programlarıyla protein yapılarındaki olası değişiklikler açısından değerlendirildi.
3.2.3Mutasyon saptanan gen bölgelerinin doğrulanması ve ebeveynlerdeki taşıyıcılığın belirlenmesi
Elde edilen DNA’lardan çalışılan gen bölgeleri PCR yöntemi ile uygun primerler (Tablo 4) kullanılarak çoğaltıldı. PCR işlemi için kullandığımız malzemeler ise; PCR Master Mix (Thermo Scientific 0.05 U/µ L), ddH2O, örenk DNA’sı ve mutasyonun olduğu bölgeyi çoğaltan primerlerdir.
Tablo 4. PCR Primerleri
Gen Ekzon Primer PCR
Ürünü (bç) Bağlanma Derecesi (ºC) BBS1 1-2 F - GTGCTCGGGCACTATTGG 550 60.2 1-2 R- CATTGTCTGACTCCCCATCC 60.3 BBS1 10-11 F- CAGAGGAGGGTCAGCCATAG 562 59.8 10-11 R- AGAAGCCCCAGAGGTGAGAC 60.8 BBS2 2 F- TGTGGATGTGTTAATAACCTGG 408 57.4 2 R- AAATAAATGATCTGATCTCTTCTAAGC 57.1 BBS4 7 F- AAACCTTGCCGAGCAATAAC 215 59.2 7 R- AACAACTTCTGCAACAAGTGC 58.1 BBS7 7 F- TGGGGAAATGTCTTATCTCTTATAGC 419 60.2 7 R- AAGGTGTGAATTGCTAAGTCTGC 59.8 10 F- AAGGGGCATGTACATTGGAG 343 59.8 10 R- AAACTTCCATTTTAAAAGGTGATAAAC 58.6 BBS9 2 F- CTTCCAGCCATTTGGATTTG 354 60.4 2 R- ACCGTGCCCAGTCTTATTTG 60 BBS10 2/2 F- TGGGATTGGTGTATTTGAGTTAG 612 58.1 2/2 R- TGTTGTTCAATGAGACCATGC 59.6 2/3 F- GATCCAAAAGATATGTTCATCTAGG 597 57.4 2/3 R- CTGGTAACATGCTTCCCTTTC 58.7 2/4 F- CCACTGCTTATTCAACAAGGG 598 59.6 2/4 R- TGACTGCTTTACTTGGCTTGA 58.7
PCR reaksiyonu için örnek sayısına göre distile su, PCR Master Mix, forward ve revers primerler (Tablo 5) hazırlandı. Her bir örnek için 0.2 ml’lik PCR tüplerine dağıtılarak, üzerlerine hastaya ait DNA örneklerinden 5 µl eklendi. ABI 2720 termal cycler ile uygun programlarda (Tablo 6) ekzonların amplifikasyonu gerçekleştirildi.
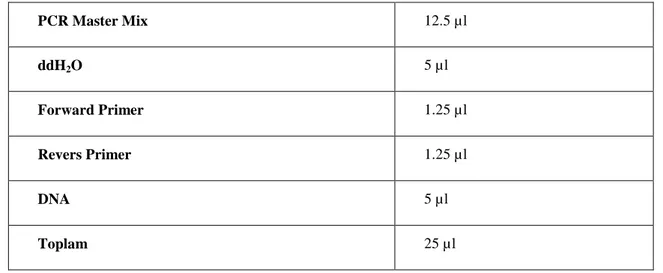
Tablo 5. Amplifikasyon İçin PCR Miksi Hazırlanışı
PCR Master Mix 12.5 µl ddH2O 5 µl Forward Primer 1.25 µl Revers Primer 1.25 µl DNA 5 µl Toplam 25 µl
Tablo 6. Ampfilikasyon İçin PCR Koşulları
Sıcaklık Süre Döngü Sayısı
Denatürasyon 94 ºC 5’ 1 94 ºC 30” 57.5 ºC 30” Denatürasyon Bağlanma Uzama 72 ºC 45” 35 Son Uzama 72 ºC 7’ 1 Bekleme 4 ºC
3.2.4 Amplifiye Edilen Bölgenin Değerlendirilmesi
PCR ürünlerinin amplifiye olup olmadığını anlamak için örneklerin yürütüleceği % 2’lik agaroz jel hazırlandı. 100 ml’lik erlen içine, 2 gr agaroz ve 100 ml 0.5XTBE konulup mikrodalga fırında ısıtılarak agarozun çözülmesi sağlandı. Çözelti berrak bir görünüm alınca, üzerine 16 µl etidyum bromür eklenerek karıştırıldı ve ürünlerin
yükleneceği kuyucukları oluşturmak için içine taraklar yerleştirilmiş tabağa döküldü. Jel donmaya bırakıldı. Parafilm üzerinde 5 µl Orange G ve 5 µl PCR ürünleri karıştırılarak kuyucuklara yüklendi. Amplifiye olan ürünlerin uzunluklarını değerlendirmek için örneklerin başı ve sonundaki kuyucuklara DNA ladder yüklendi. Örnekler 140 Volt akımda yürütüldükten sonra jel görüntüleme sistemi ile görüntüleri alındı (Şekil 2).
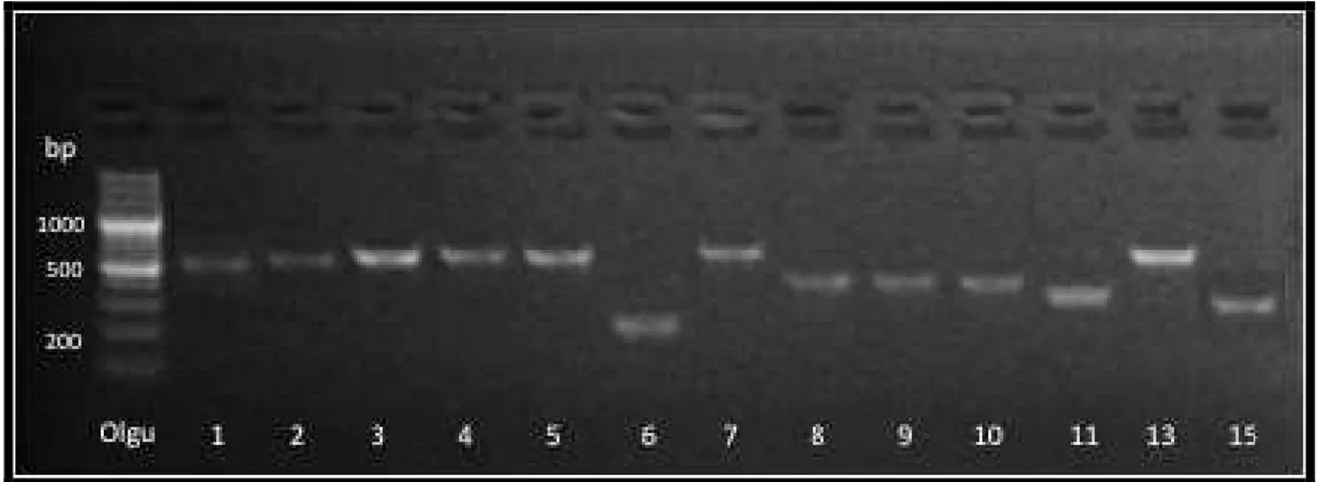
Şekil 2. BBS Genlerinin PCR Ürünleri Jel Görüntüsü
(1,4,7 ve 13 nolu bantlar BBS10 geni ekzon 2 görüntüsü, 2 nolu bant BBS1 geni ekzon 1-2 görüntüsü, 3 ve 5 nolu bantlar BBS1 geni ekzon 10-11 görüntüsü, 6 nolu bant BBS4 geni ekzon 7 görüntüsü, 8 ve 10 nolu bantlar BBS2 geni ekzon 2 görüntüsü, 9 nolu bant BBS7 geni ekzon 7 görüntüsü, 11 nolu bant BBS7 geni ekzon 10 görüntüsü, 15 nolu bant BBS9 geni ekzon 2 görüntüsüdür.)
Jel görüntüleri yorumlandıktan sonra uygun uzunlukta amplifiye olmuş ürünlere çalışmanın devam eden basamakları uygulandı.
3.2.5PCR Ürünlerinin Birinci Pürifikasyon İşlemi
Amplifiye olmuş ürünlere fazla PCR ürünlerinin ayrıştırılması için Fermentas Gene Jet PCR Purification Kit (Product No: K0701; Fermentas, USA) kiti ile aşağıdaki basamaklar izlenerek pürifikasyon işlemi yapıldı.
2. Bu işlem sonrasında PCR ürününün renginin sarıya dönmesi beklenir. Eğer renk sarıya dönmezse 10 µl 3M Sodyum Asetat eklenir.
3. PCR ürününün üzerine 20 µl İzopropanol eklendi.
4. Pipet ile karıştırıldıktan sonra önceden hazırlanmış filtreleri içerisine yerleştirilmiş receiver tüplere aktarıldı.
5. 13.000 rpm de 30 saniye santrifüj edildi. 6. Örneğin üzerine 700 µl Wash Buffer eklendi. 7. 13.000 rpm de 30 saniye santrifüj edildi. 8. Receiver tüplerinin altı boşaltıldı.
9. Filtreler tüplere tekrar konuldu ve 13.000 rpm de 1 dakika kuru santrifüj yapıldı. 10. Filtreler 1,5 ml’lik PCR tüplerine alındı.
11. Üzerine 50 µl Elution Buffer eklendi.
12. 13.000 rpm de 1 dakika santrifüj edildi ve sonrasında filtreler atıldı.
13. Tüplerde kalan örnekler Cycle Sequencing işlemine kadar 4 ºC’de saklandı.
3.2.6Pürifikasyon Ürünlerinin Cycle Sequencing İşlemi
Pürifiye edilen PCR ürünleri dideoksi veya zincir sonlanma metodu denilen yöntemle dizi analizi yapılmak üzere cycle sequencing işlemine tabi tutuldu. ABI Prism V3.1 Big-Dye Terminator Kiti (Applied Biosystems,USA), 5X Buffer (Applied Biosystems,USA), sekans primerleri ve distile su ile hazırlanan karışıma, pürifiye edilmiş ürünler eklenerek (Tablo 7) uygun PCR programında (Tablo 8) cycle sequencing yapıldı.
Tablo 7 Cycle Sequencing İçin Hazırlanan Karışım Miktar dd H2O 10.5 µl 5X Buffer 4 µl Big Dye 2 µl Primer 0.5 µl Ürün 3 µl TOPLAM 20 µl
Tablo 8. Cycle Sequencing İçin PCR Koşulları
Sıcaklık Süre Döngü Sayısı
Denatürasyon Annealing Elongasyon 96 ºC 50 ºC 60 ºC 10” 5” 4’ 25 Bekleme 4 ºC
3.2.7Ürünlerin İkinci Pürifikasyon İşlemleri
Cycle sequencing ürünlerine, içerisindeki florasan işaretli ddNTP’leri ve diğer PCR bileşenlerini ortamdan uzaklaştırmak için ticari bir pürifikasyon kiti olan Zymo Research DNA Sequencing Clean-Up Kiti (Product No: D4050; Zymo, USA) ile aşağıdaki basamaklar uygulanarak pürifikasyon işlemi yapıldı.
2. Pipet ile karıştırıldıktan sonra filtreleri içerisine yerleştirilmiş receiver tüplerin içerisine örnekler aktarıldı.
3. 13.000 rpmde 30 saniye santrifüj edildi. 4. Örneğin üzerine 300 µl Wash buffer eklendi. 5. 13.000 rpm de 30 saniye santrifüj edildi.
6. Filtreler receiver tüplerden alınarak 1,5 ml eppendorf tüplerine yerleştirildi 7. 20 µl Elution buffer eklendi
8. 13.000 rpm de 30 saniye santrifüj edildi. 9. Santrifüj sonrası filtreler atıldı.
10. Tüplerde kalan örnekler sekans cihazında yürütmek üzere platelere yüklendi.
3.2.8Örneklerin Sekans Cihazına Yüklenmesi
Platelere yüklenen örnekler, 94 ºC’de 2 dakika süreyle denatüre edilip buzda bekletildikten sonra ABI PRISM® 3130 Genetik Analizöre yüklendi. Örnekler CLC Genomics Workbench programında, http://www.ensembl.org/index.html sitesindeki BBS genlerinin dizilimi referans alınarak değerlendirildi.
4.
Bulgular
Çalışmada klinik olarak BBS ön tanısı konulan olgular alınmıştır. Yaşları 7 ay ile 21 yaş arasında değişen, 10’u erkek 5’i kadın olmak üzere, birbirleriyle akraba olmayan, 15 olgu çalışmaya dahil edilmiştir. Olguların hepsinde BBS ile ilişkili olan 16 gen (BBS1, BBS2, BBS3/ARL6, BBS4, BBS5, BBS6/MKKS, BBS7, BBS8/TTC8, BBS9, BBS10, BBS11/TRIM32, BBS12, BBS13/MKS1, BBS14/CEP290, BBS15/WDPCP, BBS16/SDCCAG8) yeni nesil dizi analizi ile çalışılmıştır. Onbeş olgunun 13’ünde hastalığa sebep olan mutasyon saptanmıştır. Mutasyonların dağılımı, BBS1 geninde 3, BBS2 geninde 2, BBS4 geninde 1, BBS7 geninde 2, BBS9 geninde 1, BBS10 geninde 4 farklı mutasyon olup tabloda ayrıntılı olarak gösterilmiştir (Tablo 9). Olguların klinik ve moleküler genetik özellikleri çalışmamızda ayrı ayrı tanımlanmıştır (Tablo 10).
Tablo 9. Çalışmada Saptanan Mutasyonlar Gen Saptandığı
olgu
Mutasyon Mutasyonun
tanımlandığı yayın
BBS1 2 HOM. IVS1-3C>G Çalışmamızda
3 HOM. Y284SfsX5 Mykytyn ve ark. 2002
5 HOM. Q338X Çalışmamızda
BBS2 8 HOM. G88AfsX6 Çalışmamızda
10 HOM. G88AfsX6 Çalışmamızda
BBS4 6 HOM. IVS6-2A>G Çalışmamızda
BBS7 9 HOM. R238EfsX59 Bin ve ark. 2009
11 HOM. L317V Çalışmamızda
BBS9 15 HOM. N35X Çalışmamızda
BBS10 1 HOM. S311A Stoetzel ve ark. 2006
4 HOM. K619IfsX10 Çalışmamızda
7 HOM. I342NfsX20 Çalışmamızda
4.1 H.A (Olgu 1)
On altı yaşında erkek olgu retinitis pigmentoza, trunkal obezite, polidaktili, küçük genitalya ve öğrenme güçlüğü nedeniyle BBS tanısı ile izlenmektedir. Olgu miadında normal spontan vajinal doğum (NSVD) ile 3500 gr. olarak dünyaya gelmiştir. Özgeçmişinde, şubat 1999’da Hirschprung hastalığı nedeniyle sağ transvers loop kolostomi operasyonu, ocak 1999’da Boley operasyonu ve kolostomi kapatma operasyonu, temmuz 1999’da postop ventral herni onarımı ve polidaktili ekstirpasyon operasyonu, sık akciğer enfeksiyonu geçirme nedeniyle yapılan tetkiklerinde Gastro özofageal reflü (GÖR) saptanması nedeniyle ekim 2000’de stapler antireflü gastroplasti ve piloroplasti operasyonu öyküsü mevcuttu. Ayrıca olgu ilki 1.5 yaşında olmak üzere 8 kez febril ve afebril nöbet geçirmiş olup 7 yaşına kadar anti epileptik ilaç kullandığı, ilaç bırakıldıktan sonra ise nöbetlerin tekrarlamadığı öğrenilmiştir. Şu anda gelişme geriliği ve konuşma bozukluğu olan olgunun ilk tek kelimeleri 2.5 yaşında söylediği fakat 7 yaşında tam konuşmanın başladığı belirtilmiştir. Olgunun bir dönem astım tanısı ile tedavi aldığı ve uyku apnesi olduğu aile tarafından bildirilmiştir.
Olgunun anne babası arasında birinci kuzen evliliği bulunmakta olup, babası ve iki halasında işitme kaybı mevcuttur (Şekil 3). Fizik muayene anındaki boy 155 cm (<3p), ağırlık 58 kg (25-50p), VKİ 24.1 kg/m2 (+0.83 SD) olarak bulundu. Olguda trunkal obezite, el ve ayaklarda brakidaktili, ellerde 5. parmakta klinodaktili, her iki ayak 2-3. parmaklar arası deri sindaktilisi, mikropenis ve geçirilmiş operasyonlara bağlı skar izi saptandı (Şekil 4). Göz muayenesi retinitis pigmentoza ile uyumlu saptanan hastanın bulgularının önce gece görme kaybı olarak başlayıp giderek ilerleyerek gündüz görmesinin de bozulduğu belirtildi. Olgunun işitme testleri normal sınırlarda saptandı. Ağız ve diş bakısında, kubbe damak, maloklüzyon ve atipik (pirinç şeklinde) diş görüldü. Yapılan IQ testi orta düzey mental retardasyon olarak yorumlandı. Psikiyatrik değerlendirmede depresyon saptandı. Rutin kardiyak değerlendirmesinde arkus aorta anomalisi saptandı, kardiyoloji takibine alındı. Ayrıca gastrik ülserine bağlı mide kanaması geçirmesi nedeniyle gastroenteroloji tarafından da takip edilmekte.
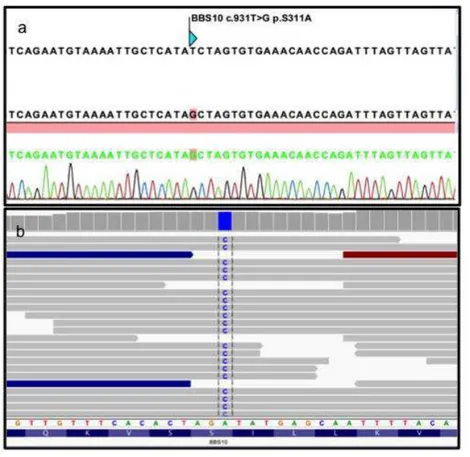
Yapılan BBS genlerinin dizi analizi sonucu BBS10 geni 2. ekzonunda homozigot 931’inci pozisyonda yer alan timin nükleotidinin yerini guanin nükleotidinin aldığı saptanmıştır. Bu değişiklik proteinin 311’inci amino asidi olan serinin yerine alaninin geçmesine (S311A) sebep olmaktadır (Şekil 5). Anne-babada değişiklik heterozigot olarak gösterilmiştir. Bu mutasyon veritabanlarında tanımlıdır.
Şekil 3. 1 No'lu Olgunun Aile Ağacı
Şekil 4. 1 No'lu Olgunun Klinik Bulguları:
Şekil 5. 1 No’lu Olgunun Sanger (a) ve NGS (b) Dizi Analizi Görüntüsü
4.2 M.Ö (Olgu 2)
Her iki tarafından birinci kuzen evliliği olan anne-babadan (Şekil 6) miadında doğan 2.5 yaşında erkek olgu obezite, polidaktili, küçük genitalya, hidronefroz ve gelişme geriliği nedeniyle BBS tanısı ile izlenmektedir. Özgeçmişinde, el parmaklarındaki polidaktilisi ve sağ tarafında inmemiş testis nedeniyle operasyon öyküsü mevcuttu. Genel gelişim basamaklarında yürüme ve tek kelimeli konuşmanın 2 yaşında başladığı öğrenildi.
Fizik muayene anındaki boy 88 cm (25-50p), ağırlık 17.5 kg (>97p), VKİ 22.6 kg/m2 (+3.28 SD) olarak bulundu. Olguda obezite, kubbe damak, el ve ayaklarda brakidaktili, ellerde 5. parmak klinodaktili, her iki ayakta post aksiyel polidaktili ve 2-3. parmaklar arası deri sindaktilisi, mikropenis, ve geçirilmiş operasyonlara bağlı skar izleri saptandı (Şekil 7). Olguda trigliserit yüksekliği saptandı ve endokrin bölümüyle izleme alındı. Batın USG sonucu hidronefroz olarak değerlendirilen olgu nefroloji takibinde olup
tedavi almamaktaydı. Psikiyatrik değerlendirmede hafif gelişme geriliği ve agresif davranış saptandı. Diğer sistem bakıları olağan saptandı.
Yapılan BBS genlerinin dizi analizi sonucu BBS1 geni 2. ekzonundan 3 önceki nükleotidin homozigot olarak sitozinden guanine (IVS1-3 C>G) yer değiştirdiği saptanmıştır (Şekil 8). Anne-babada heterozigot olarak gösterilen bu varyasyon veritabanlarında tanımlı değildir. Modelleme programları ile ekzon yapışma bölgesi değişikliği yaparak hastalık yapıcı olarak gösterildiği için patojenik bir mutasyon olduğu düşünülmektedir.
Şekil 7. 2 No'lu Olgunun Klinik Bulguları:
Dolgun yanaklar, elde 5. Parmakta klinodaktili, ayakta 2-3 deri sindaktilisi ve polidaktili
4.3 U.A (Olgu 3)
Aralarında birinci kuzen evliliği olan anne-babadan (Şekil 9) miadında NSVD ile 2200 gr doğan 16 yaşında erkek olgu obezite, görme kaybı, polidaktili, küçük genitalya ve gelişme geriliği nedeniyle BBS tanısı ile izlenmektedir. Özgeçmişinde, dört ekstremitedeki polidaktili nedeniyle operasyon öyküsü mevcuttu. Konuşma bozukluğu ve gelişme geriliği bulunan hastada, konuşmanın 5 yaşında başladığı ve gelişim basamaklarında gecikme olduğu öğrenildi. Olgunun bir dönem astım tanısı ile izlendiği öğrenildi.
Fizik muayene anındaki boy 166 cm (25-50p), ağırlık 87 kg (>97p), VKİ 31.6 kg/m2 (+2.26 SD) olarak bulundu. Olguda obezite, kubbe damak, el ve ayaklarda brakidaktili, ayakta 2-3. parmaklar arası deri sindaktilisi, mikropenis, ve geçirilmiş operasyonlara bağlı skar izleri saptandı (Şekil 10). Göz dibi bakısında periferik retinada hafif atrofi izlenen olgunun çekilen VEP sonucu bilateral kısmi ileti defekti olarak yorumlandı. Yapılan tetkiklerinde transaminaz yüksekliği, kolesterol yüksekliği ve hiperinsülinemi saptandı. Batın USG sonucu yağlı karaciğer ve hepatosplenomegali olarak değerlendirildi. Psikiyatrik değerlendirmede orta gelişme geriliği, ılımlı depresyon ve anksiyete saptandı. Diğer sistem bakıları olağan olarak değerlendirildi.
Yapılan BBS genlerinin dizi analizi sonucu BBS1 geni 10. Ekzonundaki 851. nükleotid olan adeninin homozigot delesyonu saptanmıştır. Bu değişiklik 284. pozisyondaki tirozin amino asidinin serinle yer değiştirmesine ve 5 amino asit sonra dur kodonu oluşmasına (Y284SfsX5) sebep olmaktadır (Şekil 11). Anne-babada değişiklik heterozigot olarak gösterilmiştir. Bu mutasyon veritabanlarında tanımlıdır.
Şekil 9. 3 No'lu Olgunun Aile Ağacı
Şekil 10. 3 No'lu Olgunun Klinik Bulguları: