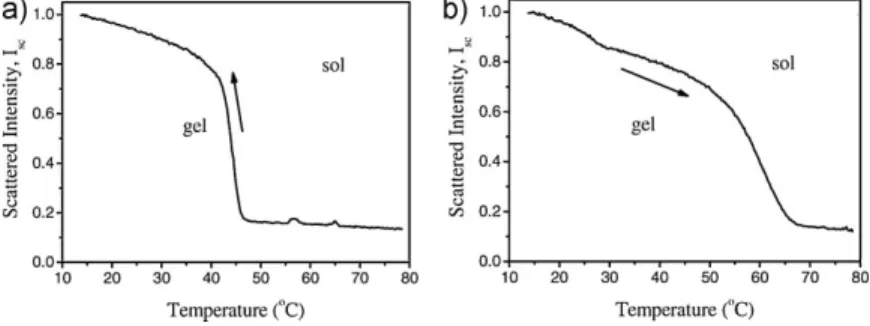
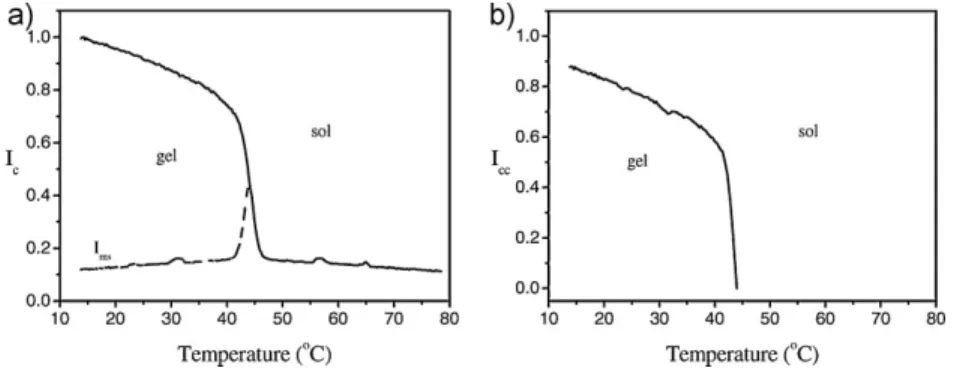
Introduction k -CarrageenaninVariousSaltSolutions CriticalExponentsofThermalPhaseTransitionsof
Tam metin
Şekil
Benzer Belgeler
Even though the effect of ammonia ratio on morphological properties of nanostructures has been well-known [28, 30] and essential fractal [31, 32] mor- phology has already
substanzların taşınmasını sağlarlar, hücre içine veya dışına geçirilecek olan moleküllerin pasif geçişine yardım ederler.. Lipid tabaka yüzeyinde bulunan proteinler
Optimization of Sol-Gel Synthesized Preceramic Polymer Precursors for Fabrication of High Purity Boron Carbide (B 4 C) Powders4. Suna AVCIOĞLU 1, 2, * , Figen KAYA 1 and Cengiz
Bu yazida spinal tümör nedeniyle ameliyat edilen ancak iki ay sonra üç kompartmanli psödomeningosel gelisen ve cerrahi olarak tedavi edilen bir olgu sunulmustur.. Olgumuz spinal
[r]
Öğretmenlerin nöbet görevleri arasında yer almayan ancak yapmaya zorlandıkları okul servis araçlarının plakasının not edilmesi; servis aracının sürücüsünün ve
DSM-5 Özgül Fobi Şiddet Ölçeği Çocuk Formunun ÇATÖ ile yapılan birlikte geçerlilik çözüm- lemesinde bağıntı katsayısı r=0.480 p<0.0001 olarak elde
Tek geçişli yakıt çevrimi; uranyumdan veya toryumdan imal edilen yakıtın reaktörde enerji üretimi için kullanılması, daha sonra kullanılmış yakıtların reaktörden
