A High-Coverage Genome Sequence
from an Archaic Denisovan Individual
Matthias Meyer,1*‡ Martin Kircher,1*† Marie-Theres Gansauge,1
Heng Li,2Fernando Racimo,1 Swapan Mallick,2,3Joshua G. Schraiber,4Flora Jay,4Kay Prüfer,1Cesare de Filippo,1 Peter H. Sudmant,6Can Alkan,5,6Qiaomei Fu,1,7Ron Do,2Nadin Rohland,2,3Arti Tandon,2,3 Michael Siebauer,1Richard E. Green,8Katarzyna Bryc,3Adrian W. Briggs,3Udo Stenzel,1 Jesse Dabney,1Jay Shendure,6Jacob Kitzman,6Michael F. Hammer,9Michael V. Shunkov,10 Anatoli P. Derevianko,10Nick Patterson,2Aida M. Andrés,1Evan E. Eichler,6,11
Montgomery Slatkin,4David Reich,2,3‡ Janet Kelso,1Svante Pääbo1‡
We present a DNA library preparation method that has allowed us to reconstruct a high-coverage (30×) genome sequence of a Denisovan, an extinct relative of Neandertals. The quality of this genome allows a direct estimation of Denisovan heterozygosity indicating that genetic diversity in these archaic hominins was extremely low. It also allows tentative dating of the specimen on the basis of“missing evolution” in its genome, detailed measurements of Denisovan and Neandertal admixture into present-day human populations, and the generation of a near-complete catalog of genetic changes that swept to high frequency in modern humans since their divergence from Denisovans.
D
raft genome sequences have been recov-ered from two archaic human groups, Neandertals (1) and Denisovans (2). Whereas Neandertals are defined by distinct morphological features and occur in the fossil record of Europe and western and central Asia from at least 230,000 until about 30,000 years ago (3), Denisovans are known only from a dis-tal manual phalanx and two molars, all exca-vated at Denisova Cave in the Altai Mountains in southern Siberia (2, 4, 5). The draft nuclear genome sequence retrieved from the Denisovan phalanx revealed that Denisovans are a sister group to Neandertals (2), with the Denisovan nuclear genome sequence falling outside Nean-dertal genetic diversity, which suggests aninde-pendent population history that differs from that of Neandertals. Also, whereas a genetic con-tribution from Neandertal to the present-day human gene pool is present in all populations outside Africa, a contribution from Denisovans is found exclusively in island Southeast Asia and Oceania (6).
Both published archaic genome sequences are of low coverage: 1.9-fold genomic coverage from the Denisovan phalanx and a total of 1.3-fold derived from three Croatian Neandertals. As a consequence, many positions in the genomes are affected by sequencing errors or nucleotide misincorporations caused by DNA damage. Pre-vious attempts to generate a genome sequence of high coverage from an archaic human have been hampered by high levels of environmental contamination. The fraction of hominin endog-enous DNA is commonly smaller than 1% and rarely approaches 5% (1, 7), which makes shot-gun sequencing of the entire genome econom-ically and logisteconom-ically impractical. The only known exception is the Denisovan phalanx, which contains ~70% endogenous DNA. However, an extremely small fragment of this specimen is available to us, and the absolute number of en-dogenous molecules that could be recovered from the sample was too low to generate high genomic coverage.
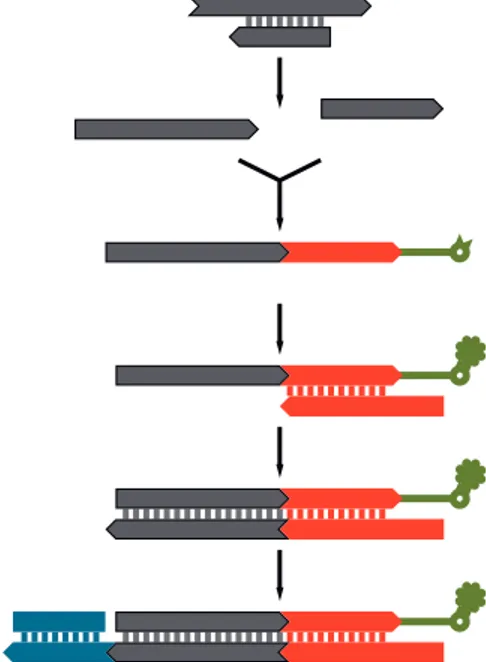
A single-stranded library preparation meth-od. DNA libraries for sequencing are normally prepared from double-stranded DNA. Howev-er, for ancient DNA the use of single-stranded DNA may be advantageous, as it will double its representation in the library. Furthermore, in a single-stranded DNA library, double-stranded molecules that carry modifications on one strand that prevent their incorporation into double-stranded DNA libraries could still be represented
by the unmodified strand. We therefore devised a single-stranded library preparation method wherein the ancient DNA is dephosphorylated, heat denatured, and ligated to a biotinylated adap-tor oligonucleotide, which allows its immobiliza-tion on streptavidin-coated beads (Fig. 1). A primer hybridized to the adaptor is then used to copy the original strand with a DNA polymerase. Fi-nally, a second adaptor is joined to the copied strand by blunt-end ligation, and the library mol-ecules are released from the beads. The entire pro-tocol is devoid of DNA purification steps, which inevitably cause loss of material.
We applied this method to aliquots of the two DNA extracts (as well as side fractions) that were previously generated from the 40 mg of bone that comprised the entire inner part of the pha-lanx (2, 8). Comparisons of these newly generated libraries with the two libraries generated in the previous study (2) show at least a 6-fold and 22-fold increase in the recovery of library mole-cules (8).
In addition to improved sequence yield, the single-strand library protocol reveals new aspects of DNA fragmentation and modification pat-terns (8). Because the ends of both DNA strands are left intact, it reveals that strand breakage occurs preferentially before and after guanine residues (fig. S6), which suggests that guanine nucleotides are frequently lost from ancient DNA, possibly as the result of depurination. It
RESEARCH ARTICLES
1
Department of Evolutionary Genetics, Max Planck Institute for Evolutionary Anthropology, D-04103 Leipzig, Germany.
2Broad Institute of Massachusetts Institute of Technology
and Harvard, Cambridge, MA 02142, USA.3Department of
Genetics, Harvard Medical School, Boston, MA 02115, USA.
4Department of Integrative Biology, University of California,
Berkeley, Berkeley, CA 94720, USA.5Department of
Com-puter Engineering, Bilkent University, 06800 Ankara, Turkey.
6Department of Genome Sciences, University of Washington
School of Medicine, Seattle, WA 98195, USA.7CAS-MPS Joint
Laboratory for Human Evolution, Institute of Vertebrate Paleon-tology and Paleoanthropology, Chinese Academy of Sciences,
100044 Beijing, China.8Jack Baskin School of Engineering,
University of California, Santa Cruz, Santa Cruz, CA 95064,
USA.9Arizona Research Laboratories, Division of
Biotechnol-ogy, University of Arizona, Tucson, AZ 85721, USA.10
Palaeo-lithic Department, Institute of Archaeology and Ethnography, Russian Academy of Sciences, Siberian Branch, 630090
Novosibirsk, Russia.11Howard Hughes Medical Institute,
Seat-tle, WA 98195, USA.
*These authors contributed equally to this work. †Present address: Department of Genome Sciences, Univer-sity of Washington, Seattle, WA 98195, USA.
‡To whom correspondence should be addressed. E-mail: [email protected] (M.M.); [email protected]. edu (D.R.); [email protected] (S.P.)
Fig. 1. For single-stranded library preparation, ancient DNA molecules are dephosphorylated and heat-denatured. Biotinylated adaptor oligonucleo-tides are ligated to the 3′ ends of the molecules, which are immobilized on streptavidin-coated beads and copied by extension of a primer hybridized to the adaptor. One strand of a double-stranded adaptor is then ligated to the newly synthesized strand. Finally, the beads are destroyed by heat to release the library molecules (not shown).
on September 28, 2017
http://science.sciencemag.org/
also reveals that deamination of cytosine resi-dues occurs with almost equal frequencies at both ends of the ancient DNA molecules. Because deamination is hypothesized to be frequent in single-stranded DNA overhangs (9, 10), this sug-gests that 5′ and 3′ overhangs occur at similar lengths and frequencies in ancient DNA.
Genome sequencing. We sequenced these libraries from both ends using Illumina’s Ge-nome Analyzer IIx and included reads for two indexes (11), which were added in the clean room to exclude the possibility of down-stream contamination with modern DNA li-braries (1). Sequences longer than 35 base pairs (bp) were aligned to the human reference ge-nome (GRCh37/1000 Gege-nome project release) and the chimpanzee genome (CGSC 2.1/panTro2) with the Burrows-Wheeler Aligner (12). After removal of polymerase chain reaction dupli-cates, genotypes were called with the Genome Analysis Toolkit (8, 13). The three Denisovan libraries yielded 82.2 gigabases of nondupli-cated sequence aligned to the human genome (8). Together with previous data (2), this pro-vides about 31-fold coverage of the ~1.86 giga-bases of the human autosomal genome to which short sequences can be confidently mapped (8). We also sequenced the genomes of 11 present-day individuals: a San, Mbuti, Mandenka, Yoruba, and Dinka from Africa; a French and Sardinian from Europe; a Han, Dai, and Papuan from Asia; and a Karitiana from South America. DNA from these individuals was bar-coded, pooled, and sequenced to ~24- to 33-fold genomic cov-erage (8). Because the samples were pooled, se-quencing errors are the same across samples and are not expected to bias inferences about population relationships.
Genome quality. We used three independent measures to estimate human contamination in the Denisovan genome sequence (8). First, on the basis of a ~4100-fold coverage of the Denisovan mitochondrial (mt) genome, we estimate that 0.35% [95% confidence interval (C.I.) 0.33 to 0.36%] of fragments that overlap positions where the Denisovan mtDNA differs from most present-day humans show the modern human variant.
Second, because the Denisovan phalanx comes from a female (2), we infer male human DNA contamination to be 0.07% (C.I. 0.05 to 0.09%) from alignments to the Y chromosome. Third, a maximum-likelihood quantification of autosom-al contamination gives an estimate of 0.22% (C.I. 0.22 to 0.23%). We conclude that less than 0.5% of the hominin sequences determined are extraneous to the bone (i.e., contamination from present-day humans).
Coverage of the genome is fairly uniform with 99.93% of the“mappable” positions covered by at least one, 99.43% by at least 10, and 92.93% by at least 20 independent DNA sequences (8). High-quality genotypes (genotype quality≥40) could be determined for 97.64% of the posi-tions. Whereas coverage in libraries prepared from ancient samples with previous methods is biased toward GC-rich sequences (14), the coverage of the libraries prepared with the single-stranded method from the Denisovan individual is similar to that of the 11 present-day human ge-nomes (prepared from double-stranded DNA), in that coverage is positively correlated with AT-content (fig. S12).
To estimate average per-base error rates in Denisovan sequence reads, we counted differ-ences between the sequenced DNA fragments and regions of the human genome that are highly conserved within primates [~5.6 mil-lion bases, (8)]. The error rate is 0.13% for the Denisovan genome, 0.17 to 0.19% for the genome sequences from the 11 present-day humans, and 1.2 to 1.7% for the two trios sequenced by the 1000 Genomes Pilot project (table S11). The lower Denisovan error rate per read is likely due to consensus-calling from duplicated reads representing the same DNA fragments and from overlap-merging of paired-end reads.
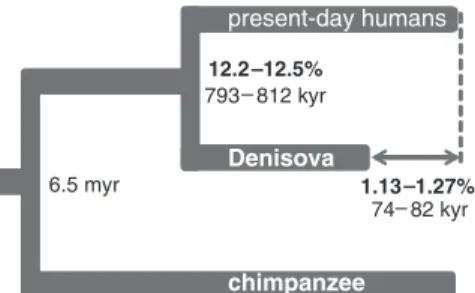
Molecular estimates of divergence and fos-sil age. We estimated the average DNA sequence divergence of all pair-wise combinations of the Denisovan genome and the 11 present-day humans as a fraction of the branch leading from the human-chimpanzee ancestor to present-day humans (Fig. 2) (8). If one assumes a human-chimpanzee av-erage DNA sequence divergence of 6.5 million years ago (15), the Denisova–present-day human divergence is ~800,000 years, close to our pre-vious estimate (2).
We next estimated the divergence of the ar-chaic and modern human populations, which must be more recent than the DNA sequence divergence. To do this, we identified sites that are variable in a present-day west African indi-vidual, who is not affected by Denisovan or Ne-andertal gene flow, and counted how often the Denisovan and Neandertal genomes carry de-rived alleles not present in chimpanzee (1). From this, we estimate the population divergence be-tween Denisovans and present-day humans to be 170,000 to 700,000 years (8). This is wider than our previous estimate (1), largely because it takes into account recent studies that broaden
the range of plausible estimates for human mu-tation rates and thus the human-chimpanzee di-vergence date.
When comparing the number of substitutions inferred to have occurred between the human-chimpanzee ancestor and the Denisovan and present-day human genomes, the number for the Denisovan genome is 1.16% lower (1.13 to 1.27%) (Fig. 2) (8). This presumably reflects the age of the Denisovan bone, which had less time to accumulate changes than present-day humans. Assuming 6.5 million years of sequence diver-gence between humans and chimpanzees, the shortening of the Denisovan branch allows the bone to be tentatively dated to between 74,000 and 82,000 years before present, in general agree-ment with the archaeological dates (2). However, we caution that multiple sources of error may affect this estimate (8). For example, the numbers of substitutions inferred to have occurred to the present-day human sequences vary by up to one-fifth of the reduction estimated for the Denisovan bone. Nevertheless, the results suggest that in the future it will be possible to determine dates of fossils based on genome sequences.
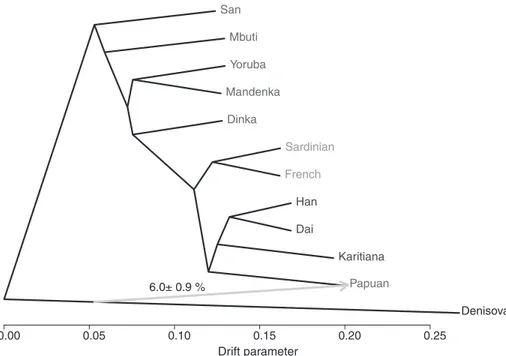
Denisovan and Neandertal gene flow. To vi-sualize the relationship between Denisova and the 11 present-day humans, we used TreeMix, which simultaneously infers a tree of relation-ships and“migration events” (16) (Fig. 3). This method estimates that 6.0% of the genomes of present-day Papuans derive from Denisovans (8). This procedure does not provide a perfect fit to the data, for example, it does not model Ne-andertal admixture. An alternative method that incorporates Neandertal admixture yields an es-timate of 3.0T 0.8% (8). This agrees with our previous finding that Denisovans have contributed to the genomes of present-day Melanesians, Aus-tralian aborigines, and other Southeast Asian is-landers (2, 6).
We tested whether Denisovans share more derived alleles with any of the 11 present-day humans (8). To increase the power to detect gene flow, we used a new approach,“enhanced” D-statistics, which restricts the analysis to alleles that are not present in 35 African genomes and are thus more likely to come from archaic humans. This confirms that Denisovans share more al-leles with Papuans than with mainland Eurasians (Fig. 4A and table S24). However, in contrast to a recent study proposing more allele-sharing be-tween Denisovans and populations from south-ern China, such as the Dai, than with populations from northern China, such as the Han (17), we find less Denisovan allele-sharing with the Dai than with the Han (although nonsignificantly so, Z = –0.9) (Fig. 4B and table S25). Further anal-ysis shows that if Denisovans contributed any DNA to the Dai, it represents less than 0.1% of their genomes today (table S26).
It is interesting that we find that Denisovans share more alleles with the three populations from eastern Asia and South America (Dai, Han, and Karitiana) than with the two European present-day humans chimpanzee Denisova 6.5 myr 12.2 12.5% 793 812 kyr 1.27% 1.13 74 82 kyr
Fig. 2. Average sequence divergence and branch length differences between the Denisovan genome and 11 present-day humans represented as a tree. Divergence is reported as fraction of the branch leading from human to the common ancestor with chimpanzee, and in years, if one assumes a human-chimpanzee divergence of 6.5 million years ago.
on September 28, 2017
http://science.sciencemag.org/
populations (French and Sardinian) (Z = 5.3). However, this does not appear to be due to Denisovan gene flow into the ancestors of present-day Asians, because the excess archaic material is more closely related to Neandertals than to Denisovans (table S27). We estimate that the proportion of Neandertal ancestry in Europe is 24% less than in eastern Asia and South America (95% C.I. 12 to 36%). One possible explanation is that there were at least two independent Ne-andertal gene flow events into modern humans (18). An alternative explanation is a single Ne-andertal gene flow event followed by dilution of the Neandertal proportion in the ancestors of
Europeans due to later migration out of Africa. However, this would require about 24% of the present-day European gene pool to be derived from African migrations subsequent to the Ne-andertal admixture.
Notably, Papuans share more alleles with the Denisovan genome on the autosomes than on the X chromosome (P = 0.01 by a two-sided test) (table S28). One possible explanation for this finding is that the gene flow into Papuan ancestors involved primarily Denisovan males. Another explanation is population substructure combined with predominantly female migration among the ancestors of modern humans as they
encountered Denisovans (which would have di-luted the Denisovan component on chromosome X) (19). A third possibility is natural selection against hybrid incompatibility alleles, which are known to be concentrated on chromosome X (20). We note that some autosomes (e.g., chro-mosome 11) also have less Denisovan ancestry (table S30), which suggests that factors such as hybrid incompatibility may be at play.
Denisovan genetic diversity. The high qual-ity of the Denisovan genome allowed us to measure its heterozygosity, i.e., the fraction of nucleotide sites that are different between a per-son’s maternal and paternal genomes (Fig. 5A). Several methods indicate that the Denisovan heterozygosity is about 0.022% (8). This is ~20% of the heterozygosity seen in the Africans, ~26 to 33% of that in the Eurasians, and 36% of that in the Karitiana, a South American population with extremely low heterozygosity (21). Because we find no evidence for unusually long stretches of homozygosity in the Denisovan genome (8), this is not due to inbreeding among the im-mediate ancestors of the Denisovan individual. We thus conclude that the genetic diversity of the population to which the Denisovan indi-vidual belonged was very low compared with that of present-day humans.
To estimate how Denisovan and modern hu-man population sizes have changed over time, we applied a Markovian coalescent model (22) to all genomes analyzed. This shows that present-day human genomes share similar population-size changes, in particular a more than twofold increase in size before 125,000 to 250,000 years ago (depending on the mutation rates assumed) (23) (Fig. 5B). Denisovans, in contrast, show a drastic decline in size at the time when the mod-ern human population began to expand.
A prediction from a small ancestral Denisovan population size is that natural selection would
Fig. 4. (A) Sharing of de-rived alleles among present-day humans, Denisovans, and Neandertals. We com-pare all possible pairs of 11 present-day humans {H1,H2} in theirD-statistics, which measure the rate at which they share derived alleles with Denisovans (x axis) and Neandertals (y axis). Each point reports T1 standard error bars from a block jackknife. D-statistics are color-coded by geographic region (“East” and“West” refer to Eurasia). Note that theD-statistic is
not the same as the mixture proportion, as it is also affected, for exam-ple, by the amount of genetic drift that is shared between the samples. (B) Sharing of derived alleles that are absent in Africans among present-day humans, Denisovans, and Neandertals. We enhance the power of the D-statistics by restricting the analysis to sites where 35 sub-Saharan Africans
have the ancestral allele and by pooling modern humans by region (bars again give one standard error). Eastern non-African populations have significantly more archaic ancestry than Western populations (Z = 5.3 and Z = 4.8 for the tests based on the Denisovan and Neandertal D-statistics, respectively). Papuan / West Papuan / East East / West -3% 0% 3% 6% 9% -3% 0% 3% 6% 9% Papuan / African Papuan / West Papuan / East East / African West / African East / West Within Region
D basic (H1, H2, Denisova, Chimp) more Denisovan relatedness
more Neandertal relatedness
D basic (H 1 , H 2 , Neandertal, Chimp) 0% 5% 10% 15% 20% 25% 0% 20% 40% 60%
D enhanced (H1, H2, Denisova, Chimp) more Denisovan relatedness
more Neandertal relatedness
D enhanced (H 1 , H 2 , Neandertal, Chimp) B A Drift parameter 0.00 0.05 0.10 0.15 0.20 0.25 Han Dinka Sardinian Papuan Dai Karitiana San Denisova Yoruba French Mbuti Mandenka 6.0± 0.9 %
Fig. 3. Maximum likelihood tree relating the Denisovan genome and the genomes of 11 present-day humans, allowing one migration event (shown as a gray arrow).
on September 28, 2017
http://science.sciencemag.org/
be less effective in weeding out slightly delete-rious mutations. We therefore estimated the ra-tio of nonsynonymous substitura-tions that are predicted to have an effect on protein function to synonymous substitutions (those that do not change amino acids) in the genomes analyzed and found it to be, in Denisovans, on average 1.5 to 2.5 times that in present-day humans, de-pending on the class of sites and populations to which Denisovans are compared (Fig. 5C) (8). This is consistent with Denisovans having a smaller population size than modern humans, resulting in less-efficient removal of a deleteri-ous mutation.
Denisovan genomic features. Because almost no phenotypic information exists about Denisovans, it is of some interest that, in agreement with a previous study (24), the Denisovan individual carried alleles that in present-day humans are as-sociated with dark skin, brown hair, and brown eyes (table S58) (8). We also identified nucle-otide changes specific to this Denisovan individ-ual and not shared with any present-day human (8). However, since we have access to only a sin-gle Denisovan individual, we expect that only a subset of these would have been shared among all Denisovans.
Of more relevance may be examination of aspects of the Denisovan karyotype. The great apes have 24 pairs of chromosomes whereas
hu-mans have 23. This difference is caused by a fu-sion of two acrocentric chromosomes that formed the metacentric human chromosome 2 (25) and resulted in the unique head-to-head joining of the telomeric hexameric repeat GGGGTT. A dif-ference in karyotype would likely have reduced the fertility of any offspring of Denisovans and modern humans. We searched all DNA fragments sequenced from the Denisovan individual and identified 12 fragments containing joined repeats. By contrast, reads from several chimpanzees and bonobos failed to yield any such fragments (8). We conclude that Denisovans and modern hu-mans (and presumably Neandertals) shared the fused chromosome 2.
Features unique to modern humans. Genome sequences of archaic human genomes allow the identification of derived genomic features that became fixed or nearly fixed in modern humans after the divergence from their archaic relatives. The previous Denisovan and Neandertal ge-nomes (1, 2) allowed less than half of all such features to be assessed with confidence. The cur-rent Denisovan genome enables us to generate an essentially complete catalog of recent changes in the human genome accessible with short-read technology (26). In total, we identified 111,812 single-nucleotide changes (SNCs) and 9499 in-sertions and deletions where modern humans are fixed for the derived state, whereas the Denisovan
individual carried the ancestral, i.e., ape-like, variant (8). This is a relatively small number. We identified 260 human-specific SNCs that cause fixed amino acid substitutions in well-defined human genes, 72 fixed SNCs that affect splice sites, and 35 SNCs that affect key positions in well-defined motifs within regulatory regions.
One way to identify changes that may have functional consequences is to focus on sites that are highly conserved among primates and that have changed on the modern human lineage after separation from Denisovan ancestors. We note that, among the 23 most conserved positions affected by amino acid changes (primate conservation score of ≥0.95), eight affect genes that are associated with brain function or nervous system devel-opment (NOVA1, SLITRK1, KATNA1, LUZP1, ARHGAP32, ADSL, HTR2B, and CNTNAP2). Four of these are involved in axonal and dendritic growth (SLITRK1 and KATNA1) and synaptic transmis-sion (ARHGAP32 and HTR2B), and two have been implicated in autism(ADSL and CNTNAP2). CNTNAP2 is also associated with susceptibility to language disorders (27) and is particularly note-worthy as it is one of the few genes known to be regulated byFOXP2, a transcription factor in-volved in language and speech development as well as synaptic plasticity (28). It is thus tempting to speculate that crucial aspects of synaptic trans-mission may have changed in modern humans.
Population size (scaled in units 4µN
e
× 1
0
3)
Time (scaled in units of 2µT)
0 5 10 15 20 0.0e+00 1.5e+05 1.0e+05 0.5e+05 Reference bases Positions A B C 0 0.5 1 1.5 2 2.5 3 10-5 10-4 10-3 10-2
5−10kya 50−100kya 0.5−1Mya 5−10Mya
San Yoruba Mandenka Mbuti Dinka French Sardinian Han Dai Papuan Karitiana Denisova 0 0.5 1 1.5 2 0 0.5 1 1.5 2 2.5
Divergent non-synonymous sites
(sample 1 / sample 2)
Heterozygous non-synonymous sites (sample 1 / sample 2)
Denisova / Modern Modern / Modern Fig. 5. (A) Heterozygosity shown by the distribution of the number of bases matching the
human reference genome at sites sampled to 20-fold coverage. They axis is scaled to show the peak representing heterozygous sites in the center. (B) Inference of population size change over time using variation in the time since the most recent common ancestors across the genome. They axis specifies a number proportional to the population size Ne. Thex axis specifies time in units of divergence per base pair (along the top in years, assuming mutation rates of 0.5 × 10−9to 1.0 × 10−9per site per year). Thin red lines around the Denisovan curve represent 100 bootstraps, which show the uncertainty of the inference. (C) The small population size in Denisovans is reflected in a greater accumulation of nonsynonymous sites (normalized by the number of synonymous sites), whether measured in terms of heterozygous sites in Denisovans versus modern humans (ratio 2.0:2.5), or the accumulation of divergent sites on the Denisovan lineage divided by modern human lineages (ratio 1.5:2.0). The analysis is restricted to nonsynonymous sites predicted to have a possibly or probably damaging effect on protein structure or function.
on September 28, 2017
http://science.sciencemag.org/
Our limited understanding of how genes re-late to phenotypes makes it impossible to pre-dict the functional consequences of these changes. However, diseases caused by mutations in genes offer clues as to which organ systems particular genes may affect. Of the 34 genes with clear as-sociations with human diseases that carry fixed substitutions changing the encoded amino acids in present-day humans, four (HPS5, GGCX, ERCC5, andZMPSTE24) affect the skin and six (RP1L1, GGCX, FRMD7, ABCA4, VCAN, and CRYBB3) affect the eye. Thus, particular aspects of the phys-iology of the skin and the eye may have changed recently in human history. Another fixed differ-ence occurs inEVC2, which when mutated causes Ellis–van Creveld syndrome. Among other symp-toms, this syndrome includes taurodontism, an enlargement of the dental pulp cavity and fusion of the roots, a trait that is common in teeth of Ne-andertals and other archaic humans. A Denisovan molar found in the cave has an enlarged pulp cavity but lacks fused roots (2). This suggests that the mutation in EVC2, perhaps in conjunction with mutations in other genes, has caused a change in dental morphology in modern humans.
We also examined duplicated regions larger than 9 kilobase pairs (kbp) in the Denisovan and present-day human genomes and found the ma-jority of them to be shared (8). However, we find 10 regions that are expanded in all present-day humans but not in the Denisovan genome. Notably, one of these overlaps a segmental du-plication associated with a pericentric inversion of chromosome 18. In contrast to humans, the Denisovan genome harbors only a partial du-plication of this region, which suggests that a deletion occurred in the Denisovan lineage. How-ever, we are unable to resolve if the pericentric inversion is indeed present in Denisovans.
Implications for archaic and modern human history. It is striking that genetic diversity among Denisovans was low although they were present in Siberia as well as presumably in Southeast Asia where they interacted with the ancestors of present-day Melanesians (6). Only future research can show how wide their geographic range was at any one time in their history. However, it is likely that they have expanded from a small population size with not enough time elapsing for genetic diversity to correspondingly increase. When technical improvements such as the one presented here will make it possible to sequence a Neandertal genome to a quality comparable to the Denisovan and modern genomes, it will be important to clarify whether the temporal tra-jectory of Neandertal effective population size matches that of the Denisovans. If that is the case, it is likely that the low Denisovan diversity reflects the expansion out of Africa of a pop-ulation ancestral to both Denisovans and Nean-dertals, a possibility that seems compatible with the dates for population divergences and popu-lation size changes presented.
By providing a comprehensive catalog of fea-tures that became fixed in modern humans after
their separation from their closest archaic rela-tives, this work will eventually lead to a better understanding of the biological differences that existed between the groups. This should ulti-mately aid in determining how it was that modern humans came to expand dramatically in popu-lation size as well as cultural complexity while archaic humans eventually dwindled in numbers and became physically extinct.
References and Notes
1. R. E. Green et al., Science 328, 710 (2010). 2. D. Reich et al., Nature 468, 1053 (2010). 3. J. J. Hublin, Proc. Natl. Acad. Sci. U.S.A. 106, 16022
(2009).
4. J. Krause et al., Nature 464, 894 (2010). 5. A. Gibbons, Science 333, 1084 (2011).
6. D. Reich et al., Am. J. Hum. Genet. 89, 516 (2011). 7. H. A. Burbano et al., Science 328, 723 (2010). 8. Materials and methods are available as supplementary
materials on Science Online.
9. A. W. Briggs et al., Proc. Natl. Acad. Sci. U.S.A. 104, 14616 (2007).
10. L. Orlando et al., Genome Res. 21, 1705 (2011). 11. M. Kircher, S. Sawyer, M. Meyer, Nucleic Acids Res. 40,
e3 (2012).
12. H. Li, R. Durbin, Bioinformatics 25, 1754 (2009). 13. A. McKenna et al., Genome Res. 20, 1297 (2010). 14. R. E. Green et al., Cell 134, 416 (2008). 15. M. Goodman, Am. J. Hum. Genet. 64, 31 (1999). 16. J. Pickrell, J. Pritchard, Inference of population splits
and mixtures from genome-wide allele frequency data. Nature Precedings (2012); http://precedings.nature.com/ documents/6956/version/1.
17. P. Skoglund, M. Jakobsson, Proc. Natl. Acad. Sci. U.S.A. 108, 18301 (2011).
18. M. Currat, L. Excoffier, Proc. Natl. Acad. Sci. U.S.A. 108, 15129 (2011).
19. R. J. Petit, L. Excoffier, Trends Ecol. Evol. 24, 386 (2009).
20. J. A. Coyne, H. A. Orr, in Speciation and its Consequences, D. Otte, and J. A. Endler, Eds. (Wiley, New York, 1989), pp. 180–207.
21. J. R. Kidd, F. L. Black, K. M. Weiss, I. Balazs, K. K. Kidd, Hum. Biol. 63, 775 (1991).
22. H. Li, R. Durbin, Nature 475, 493 (2011). 23. D. F. Conrad et al., Nat. Genet. 43, 712 (2011). 24. C. C. Cerqueira et al., Am. J. Hum. Biol. 24, 705 (2012). 25. J. W. IJdo, A. Baldini, D. C. Ward, S. T. Reeders, R. A. Wells,
Proc. Natl. Acad. Sci. U.S.A. 88, 9051 (1991). 26. R. M. Durbin et al., Nature 467, 1061 (2010). 27. S. C. Vernes et al., N. Engl. J. Med. 359, 2337 (2008). 28. W. Enard et al., Cell 137, 961 (2009).
Acknowledgments: The Denisovan sequence reads are available from the European Nucleotide Archive (ENA) under study accession ERP001519. The present-day human sequence reads are available from the Short Read Archive (SRA) under accession SRA047577. Alignments and genotype calls for each of the sequenced individuals are available at www.eva.mpg.de/denisova/. In addition, the Denisovan sequence reads and alignments are available as a public data set via Amazon Web Services (AWS) at http://aws.amazon.com/datasets/2357/ and as a track in the University of California, Santa Cruz genome browser. We thank D. Falush, P. Johnson, J. Krause, M. Lachmann, S. Sawyer, L. Vigilant and B. Viola for comments, help, and suggestions; A. Aximu, B. Höber, B. Höffner, A. Weihmann, T. Kratzer, and R. Roesch for expert technical assistance; R. Schultz for help with data management; and M. Schreiber for improvement of graphics. The Presidential Innovation Fund of the Max Planck Society made this project possible. D.R. and N.P. are grateful for support from NSF HOMINID grant no. 1032255 and NIH grant GM100233. J.G.S., F.J., and M.S. were supported by NIH grant R01-GM40282 to M.S. P.H.S. is supported by an HHMI International Student Fellowship. F.R. is supported by a German Academic Exchange Service (DAAD) study scholarship. E.E.E. is on the scientific advisory boards for Pacific Biosciences, Inc., SynapDx Corp, and DNAnexus, Inc.
Supplementary Materials
www.sciencemag.org/cgi/content/full/science.1224344/DC1 Materials and Methods
Figs. S1 to S38 Tables S1 to S58 References (29–196)
7 May 2012; accepted 14 August 2012 Published online 30 August 2012; 10.1126/science.1224344
Cilia at the Node of Mouse
Embryos Sense Fluid Flow for
Left-Right Determination via Pkd2
Satoko Yoshiba,1Hidetaka Shiratori,1Ivana Y. Kuo,2Aiko Kawasumi,1* Kyosuke Shinohara,1Shigenori Nonaka,3Yasuko Asai,1Genta Sasaki,1 Jose Antonio Belo,4Hiroshi Sasaki,5Junichi Nakai,6Bernd Dworniczak,7 Barbara E. Ehrlich,2Petra Pennekamp,7,8† Hiroshi Hamada1†
Unidirectional fluid flow plays an essential role in the breaking of left-right (L-R) symmetry in mouse embryos, but it has remained unclear how the flow is sensed by the embryo. We report that the Ca2+channel Polycystin-2 (Pkd2) is required specifically in the perinodal crown cells for sensing the nodal flow. Examination of mutant forms of Pkd2 shows that the ciliary localization of Pkd2 is essential for correct L-R patterning. WhereasKif3a mutant embryos, which lack all cilia, failed to respond to an artificial flow, restoration of primary cilia in crown cells rescued the response to the flow. Our results thus suggest that nodal flow is sensed in a manner dependent on Pkd2 by the cilia of crown cells located at the edge of the node.
M
ost of the visceral organs in vertebrates exhibit left-right (L-R) asymmetry in their shape and/or position. Thebreak-ing of L-R symmetry in the embryos of many vertebrates is mediated by a unidirectional fluid flow in the ventral node (an embryonic cavity
on September 28, 2017
http://science.sciencemag.org/
Montgomery Slatkin, David Reich, Janet Kelso and Svante Pääbo
Kitzman, Michael F. Hammer, Michael V. Shunkov, Anatoli P. Derevianko, Nick Patterson, Aida M. Andrés, Evan E. Eichler, Michael Siebauer, Richard E. Green, Katarzyna Bryc, Adrian W. Briggs, Udo Stenzel, Jesse Dabney, Jay Shendure, Jacob Flora Jay, Kay Prüfer, Cesare de Filippo, Peter H. Sudmant, Can Alkan, Qiaomei Fu, Ron Do, Nadin Rohland, Arti Tandon, Matthias Meyer, Martin Kircher, Marie-Theres Gansauge, Heng Li, Fernando Racimo, Swapan Mallick, Joshua G. Schraiber,
originally published online August 30, 2012 DOI: 10.1126/science.1224344
(6104), 222-226.
338 Science
archaics, including Neandertals, and establishes a catalog of genetic changes within the human lineage.
because of a small population size. The genome sequence also illuminates the relationships between humans and evidence for very low rates of heterozygosity in the Denisova, probably not because of recent inbreeding, but instead online 30 August) provide a detailed analysis of a high-quality Denisovan genome. The genomic sequence provides
(p. 222, published
et al.
Meyer
ancestors of modern-day humans. Using a single-stranded library preparation method,
The Denisovans were archaic humans closely related to Neandertals, whose populations overlapped with the
Ancient Genomics
ARTICLE TOOLS http://science.sciencemag.org/content/338/6104/222
MATERIALS SUPPLEMENTARY http://science.sciencemag.org/content/suppl/2012/08/29/science.1224344.DC1 CONTENT RELATED http://science.sciencemag.org/content/sci/337/6098/1028.full REFERENCES http://science.sciencemag.org/content/338/6104/222#BIBL
This article cites 191 articles, 41 of which you can access for free
PERMISSIONS http://www.sciencemag.org/help/reprints-and-permissions
Terms of Service
Use of this article is subject to the
is a registered trademark of AAAS.
Science
licensee American Association for the Advancement of Science. No claim to original U.S. Government Works. The title Science, 1200 New York Avenue NW, Washington, DC 20005. 2017 © The Authors, some rights reserved; exclusive
(print ISSN 0036-8075; online ISSN 1095-9203) is published by the American Association for the Advancement of
Science
on September 28, 2017
http://science.sciencemag.org/