Z 0 2 ■ · Γ » · η · ^ · . .!·*·. * ч * > · · i . , i¡2é¿^*^í2w. ¿ w ..í , ^ ^ ií? •^^■'■•Vi. *»'"*'· ·» '::···'?,*■ »J ,«v. · .■■ N ' ^·· Л «fc i» ’ 3éf/-’<A ^ .t . * t* - -*, *· · ^. ·» ^ ^,,1·^ % ^ 'w ^ _ '· ■ ■ ‘ lit·'·· ¿ ■ ·* .r * ^'. · · í^■ .H Л'Ч· · , .■«λ·, міЫмі İM· AAWwt ν«ί«ι·ί»*ιΓ w 'M tár
Jíi. «íU^xIi:.·:. . i>i*l-V|k ««Й* Ml Jlÿ *Ч» 4«ίίΐ '»М^^ «Mi 4«^·> *'·\'·' :·^ ; і>’.ν,^,.· \ic¡? Ш ί·. ΐ;;ΐ;*"ι^'··
PRODUCTION OF RECOM BINANT HUM AN p-CATENIN
PROTEIN IN
E.coli
A N D SCREENING FOR ANTI-P-CATENIN
AUTOANTIBODIES IN CANCER SERA
A THESIS SUBM ITTED TO
THE DEPARTMENT OF MOLECULAR BIOLOGY AND
GENETICS AND THE INSTITUTE OF ENGINEERING AND
SCIENCE OF BILKENT UNIVERSITY
IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR
THE DEGREE OF M ASTER OF SCIENCE
By
Nuri Oztürk
о?: 2о2
'09?
I certify that I have read this thesis and that, in my
opinion, it is fully adequate, in scope and in quality,
as a dissertation for the degree of Master of Science.
Prof Dr. Mehmet Öztürk
I certify that I have read this thesis and that, in my
opinion, it is fully adequate, in scope and in quality,
as a dissertation for the degree of Master of Science.
Prof. Dr. Meral Özgüç
I certify that I have read this thesis and that, in my
opinion, it is fully adequate, in scope and in quality,
as a dissertation for the degree of Master of Science.
Assist. Prof Rengul Qetin-Atalay
Approved for the Institute of Engineering and Science.
Prof Dr. Mehmet Baray
Director of Institute of Engineering and Science
ABSTRACT
PRODUCTION OF RECOMBINANT HUMAN P-CATENIN PROTEIN BSf E.coli AND
SCREENING FOR ANTI-p-CATENIN AUTO-ANTIBODIES IN CANCER SERA
Nuri Ôztürk
M.S in Molecular Biology and Genetics Supervisor; Prof. Dr. Mehmet Ôztürk
July 2000
The p-catenin gene is a component of wnt pathway involved in developmental pattern formation. The protein product is involved in both cell-cell interaction through E- cadherin and gene regulation through Tcf transcription fectors. In the absence of wnt signaling, P-catenin protein is degraded actively following phosphorylation of serine/threonine residues at its N-terminal region. In many cancer types (colorectal cancer, hepatocellular carcinoma, hepatoblastoma, melanoma, thyroid cancers...), P- catenin protein accumulates aberrantly due to a loss of active degradation process. In colorectal cancers, this is due to APC mutations, in others p-catenin mutations occur at or around exon 3 leading to a loss of degradation box of the protein product. It is believed that accumulated p-catenin protein result in increased cell growth by transcriptional activation of genes such as c-myc and cyclin D. A similar aberration was also observed with p53 gene whose mutations lead to the accumulation of mutant p53 proteins and loss of growth control. In addition, cancer patients expressing mutant p53 protein in their tumor cells develop anti-p53 autoantibodies, as well as cytotoxic T cell (CTL) response. Anti p-catenin CTL response in melanoma patients has been reported, but it is unknown whether cancer patients also develop anti-P-catenin autoantibodies.
Our aim was to test whether cancer patients develop anti-P-catenin autoantibodies. For this aim, we first produced an N-terminally truncated recombinant P-catenin protein lacking the first 88 amino acid residues. The corresponding p-catenin cDNA was subcloned into an E. coli expression vector (pQE-32) with a bxHistidine tag at N-terminus. Affinity purified recombinant P-catenin protein was used as antigen to screen cancer patient and control sera by different immunoassays. Initially, more than 1500 sera were screened by an ELISA method and 53 sera were selected for further studies. The presence of anti-P-catenin autoantibodies was confirmed in 5 patients by western blot assay. All of these sera also reacted with in vitro produced full length catenin protein using immunoprécipitation assay. Five control sera were negative in this sensitive assay. This work demonstrated that some cancer patients develop auto antibodies to P-catenin. Such auto-antibodies may serve as tumor markers for patients displaying P-catenin mutations. Further studies are needed to determine the frequency and the specificity of anti p-catenin autoantibody response in different tumor types.
ÖZET
E.COLİ’DE REKOMBINANT İNSAN BETA-KATENİN PROTEİNİNİN ÜRETİMİ VE KANSER SERUMLARINDA BETA-KATENİN OTO ANTİKORLARININ
TARANMASI
Nuri Öztürk
Moleküler Biyoloji ve Genetik Yüksek Lisansı Tez Yöneticisi: Prof Dr. Mehmet Öztürk
Temmuz 2000.
P-katenin geni, gelişimsel doku biçimlenmesinde rol oynayan wnt yolağının bir parçasını oluşturmaktadır, p-katenin proteini hem E-kaderin aracılığı ile hücreler arası etkileşimde, hem de Tef transkripsiyon faktörleri aracılığı ile gen ekspresyonunun düzenlenmesinde yer alan bir proteindir. Wnt uyarısı olmadığı zaman, hücrelerdeki P-katenin proteini N- terminal bölgesinde yer alan serin/treonin amino asitlerinin fosforlanması sonucu, aktif olarak yıkılır. Bir çok kanserde (kolorektal kanser, hepatoselüler kanser, hepatoblastoma, melanoma, tiroid kanseri vb.), aktif yıkılım sistemindeki anormallikler nedeni ile, P- katenin proteini hücrelerde aşırı düzeyde birikim gösterir. Kolorektal kanserlerde bu birikimin nedeni APC geni mutasyonlarıdır. Diğer kanserlerde ise, P-katenin proteinin yıkılım bölgesini kodlayan 3. ekzon ve çevresinde oluşan mutasyonlar bu birikime neden olmaktadır. Kanserli hücrelerde biriken P-katenin proteininin, c-myc ve eyelin D gibi genlerin transkripsiyonel aktivasyonuna neden olarak, aşın hücre çoğalmasına yol açtığına inanılmaktadır. Benzer bir anormallik, mutant p53 proteininin hücre çoğalmasına yolaçtığı p53 geni mutasyonlarmda da gözlemlenmiştir. Buna ek olarak, tümörlerinde mutant p53 proteini eksprese eden kanserli hastaların p53 proteinine karşı otoantikor ve sitotoksik T hücresi (CTL) yanıtı geliştirdikleri gösterilmiştir. Bazı melanomalı hastalarda P-katenine karşı CTL yanıtı gösterilmiştir, ancak kanserli hastaların anti p- katenin oto antikoru geliştirip-geliştirmedikleri bilinmemektedir.
Kanserli hastaların P-katenine karşı otoantikor geliştirip-geliştirmediklerini test etmek amacıyla, önce N-terminal tarafından kısaltılmış (ilk 88 amin asidi eksik) insan p- katenin proteini rekombinant olarak üretildi. İlgili p-katenin cDNA sı E. Coli ekspresyon vektörü olan pQE-32 plazmidine N-terminalinden 6xHistidin işaretli olarak klonlandı. Affinité kromatografisi ile saflaştırılan rekombinant p-katenin protenin kanserli hasta ve kontrol serumlarının, farklı immünolojik testlerle taranmasında antijen olarak kullanıldı. İlk aşamada, 1500 den fazla serum ELİSA yöntemi ile tarandı ve 53 kanserli hasta serumu seçildi. Bu serumlardan 5 adedinde anti P-katenin otoantikoru varlığı western immunoblot yöntemi ile gösterildi. Bu 5 serum in vitro olarak üretilmiş normal boyda P- katenin proteini ile yapılan immunoçöktürme yöntemi ile de pozitif sonuç verdi. Buna karşılık, 5 kontrol serumu bu duyarlı yöntemle negatif sonuç verdi. Bu çalışma bazı kanserli hastaların p-katenine karşı otoantikor geliştirdiklerini göstermiştir. Değişik tümörlerde anti P-katenin otoantikor yanıtının oranı ve özgünlüğünü belirlemek için ek çalışmaların yapılması gerekmektedir.
ACKNOWLEDGEMENT
Thank you ...
... Dr. Mehmet Öztürk, my supervisor, for all that I have learnt from you and for
your patience.
... Dr. Rengül Çetin-Atalay, for the knowledge and experience you have shared
and the support you have given.
Tolga Çağatay for the knowledge and experience you have shared.
Gülayşe İnce for the knowledge you taught and experience you have shared.
Emre and Berna Sayan for the knowledge you have shared and the support you
have given.
.. .Esra Yıldız for your help.
TABLE OF CONTENTS
SIGNATURE PAGE...iii
ABSTRACT... iv
ÖZET... V ACKNOWLEDGEMENTS... vi
TABLE OF CONTENTS... vii
LISTOF FIGURES... xii
LIST OF TABLES... xiv
ABBREVIATIONS... xv
CHAPTER 1. INTRODUCTION
1.1.1 A quick review on cancer gene products 1
1.1.2 Human p-catenin gene; C77WS7 2
1.1.3 P-catenin protein 3
1.1.4 p-catenin in cell-adhesion complex 5
1.1.5 P-catenin as a bi-functional protein 7
1.1.6 Wnt signaling 8
1.1.7 Genes activated by Wnt signal 10
1.2.1 Mutations of P-catenin gene 12
1.2.2 Mutations of P-catenin gene in experimentally induced animal tumors 17
1.2.3 The accumulation of P-catenin protein in tumor cells 18
1.3 Tumor antigens, tumor associated-antigens and autoantibodies 20
1.3.1 T umor antigens 20
1.3.2 T umor-associated antigens (T AA) 21
1.3.3 Autoantibodies in tumor hosts 22
1.3.4 Cytoxic T cell (CTL) responses against p53 and P-catenin 25
1.4 AIM AND STRATEGY 28
CHAPTER 2. MATERIAL AND METHODS
2.1 MATERIALS 29 2.1.1 Chemicals 29 2.1.2 Bacterial strains 29 2.1.3 Enzymes 29 2.1.4 Electrophoresis 29 2.1.5 Antibodies 29 2.1.6 Kits 30
2.1.7 DNA and Protein size markers 30
2.2 SOLUTIONS AND MEDIA 31
2.2.1 Agarose gel electrophoresis (AGE) solutions 31
2.2.2 Solutions for plasmid DNA isolation (mini-prep) 31
2.2.3 Solutions for bacterial transformation 31
2.2.4 Microbiological media and antibiotics 31
2.2.5 Polyacrylamide gel electrophoresis solutions 32
2.2.6 Protein purification buffers 33
2.2.7 Solutions for protein concentration estimation 33
2.2.8 ELISA and Western Blotting solutions and reagents 33
2.2.9 IP buffers 34
2.3 METHODS 36
2.3.1 Transformation of bacterial cells 3 6
2.3.2 Preparation of competent cells 36
2.3.2.1 Conventional “Calcium Chloride” method 36
2.3.2.2 Super competent Æ. Co//preparation 36
2.3.3 Transformation 37
2.3.3.1 Conventional “Calcium Chloride “ transformation 3 7
2.3.3.2 Super competent E'.co//transformation 38
2.3.4 Long term storage of bacterial strains 39
2.3.5 Small scale preparation of plasmid DNA (Construct) (mini-prep) 38
2.3.6 Midiprep of plasmids or constmcts 40
2.3.7 Quantification of double-stranded DNA 40
2.3.8 Restriction analysis and enzyme digestion of plasmid DNA (construct) 40
2.3.9 Extraction and precipitation of DNA 41
2.3.9.1 Phenol extraction 41
2.3.9.2 Ethanol precipitation 41
2.3.10 Agarose gel electrophoresis of DNA 41
2.3.11 Recovery of DNA fragments from agarose gel 42
2.3.12 Ligation of DNA fragments 42
2.3.13. Expression of subcloned fragment 42
2.3.13.1 Rapid screening of small-scale expression cultures 44
2.3.13.2 Checking for solubility of the expressed protein 44
2.3.13.3 Denaturing purification of insoluble proteins 45
2.3.14 Dialysis 46
2.3.15 Concentrating the purified proteins
2.3.16 Screening of sera for autoantibodies
2.3.17 ELISA (Enzyme-Linked ImmunoSorbant Assay)
2.3.18 SDS Polyacrylamide gel electrophoresis
2.3.19 Immunological detection of immobilized proteins (Western Blotting)
2.3.19.1 Transfer of proteins onto membranes
2.3.19.2 Immunological detection of the transferred proteins
2.3.20 In vitro Transcription-Translation (IVTT) reactions
2.3.21 Immunoprécipitation CHAPTER 3. RESULTS 46 47 48 49 49 49 50 51 53 46
3.1.1 Subcloning of 2.08 kb fi'agment of P-Catenin into pQE-32 bacterial
expression system 53
3.1.2 Transformation of DH5a bacteria by pQE32-iS(afc I-P-catenin-<Sa/1 57
3.1.3 Rapid Screening of small-scale expression of recombinant P-catenin
fragment 58
3.1.4 Checking for protein solubility 60
3.1.5 Denaturing purification of insoluble recombinant P-catenin protein 61
3.1.6 Concentrating the purified P-Catenin 63
3.1.7 Optimization of ELISA with monoclonal antibodies 64
3.1.8 Screening of cancer patient and control sera for P-catenin autoantibodies using
ELISA 65
3.1.8 Western blotting results
3.1.9 rVTT and Immunoprécipitation of P-catenin
66
Summary of results CHAPTER 4. DISCUSSION Future Perspectives REFERENCES APPENDICES 72 73 75 77 88 XI
LIST OF FIGURES
Figure 1; p-catenin protein and its interaction regions and degradation box 4
Figure 2: P-catenin in adherens-junctions 6
Figure 3; A summary of P-catenin functions 10
Figured: Hot spots for mutations in p-catenin 14
Figure 5: Deletions in p-catenin gene and the deleted amino acids in the
protein 16
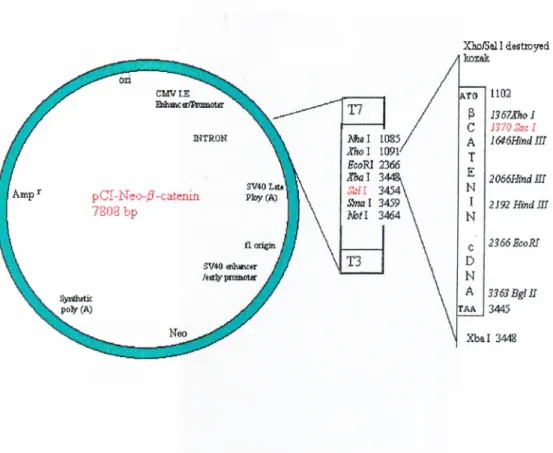
Figure 3-1: Map of pCI-Neo-p-catenin mammalian expression vector. 54
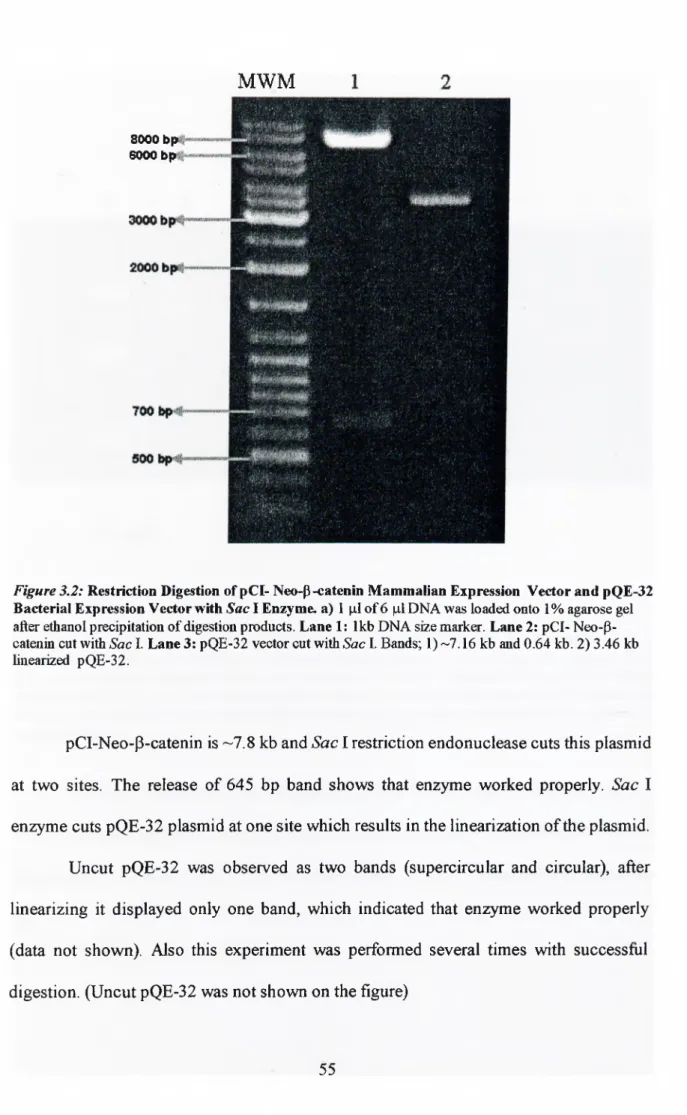
Figure 3-2: Restriction digestion of pQE- Neo-P-catenin mammalian expression
vector and pQE-32 bacterial expression vector with Sac I enzyme 55
Figure 3-3: Restriction digestion with Sal I following Sac I digestion 56
Figure 3-4: Strategy of subcloning. 57
Figure 3-5: Rapid screening of recombinant protein expression. 59
Figure 3-6: Immunoblotting of bacterial lysates 60
Figure 3-7: Solubility of recombinant p-catenin as tested by comassie blue staining
61
Figure 3-8: Purification of recombinant P-catenin from insoluble fraction of Ml 5-12a
lysates 62
Figure 3-9: Dialyzed and concentrated protein fractions and estimation of protein
concentration 64
Figure 3-10: Determination of the most suitable concentration of antigens for ELISA
with monoclonal antibodies 65
Figure 3-11: Determination of p53 saturation point by antibodies 67
Figure 3-12: Determination of P-catenin saturation point by anti-P-catenin monoclonal
antibody
Figure 3-13: Screening of cancer sera
Figure 3-14: Confirmation of p-catenin autoantibody positive sera.
Figure 3-15: IVTT and IP of p-catenin
68
69
70
71
LIST OF TABLES
Table 1:
Table 2:
Table 3;
Somatic mutations of P-catenin human cancers 12
Mutation spectra of P-catenin gene in human cancers 13
Number of the mutations detected in P-catenin (between a.a. 32 and
50 of exon 3) 15
ABBREVIATIONS
a. a. amino acid
A^oo absorbance at 600-nanometer wavelength
Ab Antibody
AFP Alpha feto-protein
AGE agarose gel electrophoresis
APC adenomatous polyposis coli
APS ammonium persulfate
Arg Arginine
Arm Armadillo
bp base pair
BSA Bovine serum albumin
CEA Carcinoembryonic antigen
cDNA complementary DNA
CIA carcinoma in adenoma
cm centimeter
CR Colorectal cancer
CRD cysteine-rich domain
CTL cytotoxic T lymphocyte
ddHzO double distilled water
DEN Diethyl nitrosamine
DNA deoxyribonucleic acid
Dsh Dishevelled
EDTA ethylenediamine-tetra-acetic acid
Fz Frizzled
x g gravity
g gram
GSK-3P Glycogen synthase kinase 3P
HB hepatoblastoma HCC Hepatocellular carcinoma hr hour HRP horseradish peroxidase He Isoleucine n> Immunoprécipitation n>TG isopropylthio-beta-D-galactoside
r/T T in vitro transcription and translation
kb kilo base
kDa kilo Dalton
LB Luria-Bertani Medium
M molar
mAb monoclonal antibody
MAM Methylazoxymethanol
ME mercaptoethanol
Met Methionine
min minute
MidiPrep medium-scale isolation of plasmid DNA by
MiniPrep small-scale isolation of DNA by alkaline lys
ml milliliter
mRNA messenger RNA
mt mutant type
nm nanometer
NTA nitrilo-tri-acetic-acid
PAGE polyacrylamide gel electrophoresis
Pan pangolin
PBS phosphate buffered saline
PCR polymerase chain reaction
PVDF polyvinilydene difluoride
Pore Porcupine
rpm revolution per minute
RNA ribonucleic acid
RNase ribonuclease
SDS sodium dodecyl sulphate
sec second
Ser Serine
SSHN Squamous cell carcinoma of head and neck
TAE tris/acetic acid/EDTA buffer
Tcf T cell fector
TIL Tumor-infiltrating lymphocyte
TEMED N,N,N,N-tetramethyl-l ,2 diaminoethane
Tris 2-amino-2-[hydroxymethyl]-l,3 propandiol
Tyr tyrosine
U unit
V volt
v/v volume for volume
WB Western immunoblotting
Wg Wingless
Wt wild type
w/v weight for volume
w/w weight for weight
Zw3 zeste-white 3
CHAPTER 1 INTRODUCTION 1.1.1 A Quick Review on Cancer Gene Products
Tumors result from a deficiency in the mechanisms controlling the normal
growth, location and mortality of cells in multi-cellular organisms. The genes in which
acquisition of mutations may trigger tumor development can be addressed as tumor genes
and their products as tumor gene products. The mutations triggering tumor development
arise in proto-oncogenes, tumor suppressor genes, and genes encoding DNA repair
proteins (Pegg 1999).
The proto-oncogenes encode the normal components of signaling pathways
which control cell proliferation. The mutated forms of proto-oncogenes, which are named
then as “oncogenes”, may escape regulation of cell growth. Oncogenes were originally
identified in viruses. These vimses may induce tumors in animals and/or transform cells
in vitro. The oncogenes carried by these viruses are strongly homologous to some
sequences involved in normal cellular signaling pathways, such as c-Myc in animals
versus v-myc in virus. As another example of oncogenes, Wnt-1, a member of the Wnt
family genes, was originally identified as an oncogene activated by the insertion of
mouse mammary tumor vims (MMTV) into host DNA in vims-induced mammary
adenocarcinomas (Nusse et al. 1984). In the downstream of Wnt pathway (reviewed by
Cadigan and Nusse 1997), ß-catenin was recognized as another proto-oncogene. It is
believed that accumulation of ß-catenin induces uncontrolled activation of downstream
genes, some of which might be involved in tumorigenesis. Among others, Cyclin D1 and
c-Myc are two oncogenes, recently shown to be up regulated by mutant ß-catenin (He et
“Tumor suppressor genes”, the loss of function of which leads to deregulated
control of cell cycle progression, cellular adhesion etc., generally exhibit a recessive
behavior. As an example of tumor suppressor genes aberrations, alterations in the tumor
suppressor/>55 gene are involved in the genesis of diverse types of human malignancies
(reviewed by Ko and Prives 1996). At least 50% of all malignant tumors have
abnormalities in the p53 gene, which is located in chromosome 17 (reviewed by Souissi
T., Y. Legros 1994). On cellular stress, especially induced by DNA damage, p53 can
arrest cell cycle progression to allow DNA repair (Kastan et al. 1991) or can lead to
programmed cell death, also called apoptosis (Yonish-Rouach et al. 1991). Mutated p53
accumulates in cells, since the decreased rate of mutated p53 degradation results in the
longer half-life of the protein (Kupryjanczyk et al. 1993). Another tumor suppressor is
APC (Adenomatous polyposis coli), which forms dimers via the leucine zipper region
and associates with p-catenin (reviewed by Polakis 1997).
The third group genes in which mutations may trigger tumorigenesis are DNA
repair genes. Mutations in these enzymes promote genetic instability.
1.1.2 Human p-Catenin Gene: CTNNBl
P-catenin was first isolated as a protein associated with the intracellular domain of
E-cadherin (McCrea et al. 1991). Nollet et al. determined the primary structure of the
human p-catenin gene (CTTNBl) by analysis of cDNA and genomic clones. The size of
the complete human P-catenin gene was determined to be 23.2 kb. P-catenin consists of
16 exons; sizes of which range from 61 to 790 bp. Half of the introns are smaller than
3p22-p21.3 (Kraus et al. 1994). The house mouse (Mus musculus) P-catenin gene is
named as Catnb.
Nollet et al. 1996, showed a major transcription initiation as an “A” nucleotide at
—214 to the ATG initiation codon. Transcription product of p-catenin gene is 3362
nucleotides long. As a result, Nollet et al. compiled a complete mRNA sequence of 3362
bp; 214 of 5’UTR, 2346 bp of coding sequence, and 802 bp of 3’UTR. An alternatively
spliced form of transcript is reduced by 159 bp at 3’UTR in exon 16. They found the two
alternatively spliced forms of human p-catenin mRNA at equal ratio in 14 human cell
lines by RT-PCR. The 5’ flanking region of CTNNBl is highly GC-rich and contain a
TATA box, as well as putative binding sites for some transcription fectors, such as
NFKB, SPl, AP2, andEGFRl (Nolletera/. 1996)
1.1.3 P-Catenin Protein
Although, p-catenin was first isolated in association with E-cadherin cytoplasmic
domain, this protein is an essential component of the WntAVingless (also called wnt or
Wg) signaling pathway. Wnt pathway is involved in developmental processes, such as
segment polarity determination in Drosophila, dorso-ventral patterning (axis
specification) in early Xenopus development, and mesoderm induction in C. elegans
(reviewed by Cadigan and Nusse 1997).
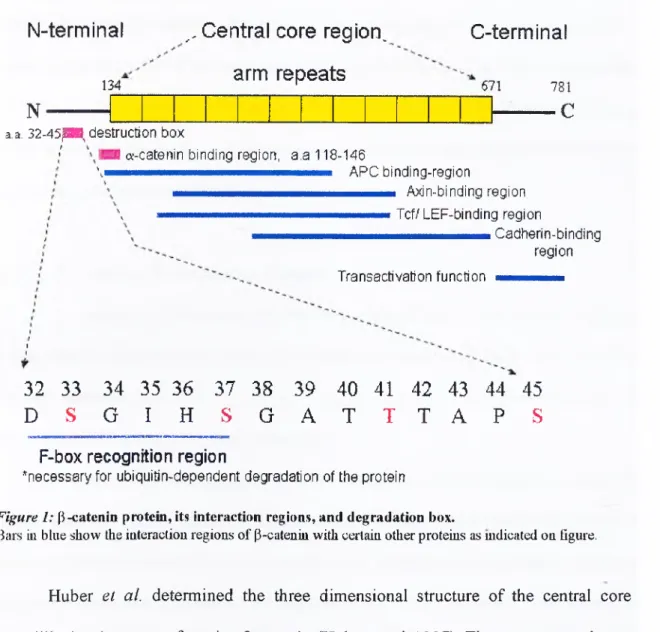
The P-catenin protein is composed of an amino-terminal region of 134 amino
acids, a central core region (12-imperfect 42-a.a. armadillo, arm, repeats) o f 550 a.a., and
a carboxy-terminal region (including a trans-activation domain) of 100 a.a. In the amino
45 S) acting as phosphorylation sites (Figure 1). Glycogen synthase kinase-3 P (GSK-3P)
phosphorylâtes P-catenin at these sites, to generate a tag for degradation of p-catenin by
ubiquitination-proteosome pathway. The ubiquitination of the protein is necessary for its
rapid turnover in the cells (reviewed by Willertand Nusse 1998).
N-terminal
134
^.Central core region^ arm repeats C-terminal 671
N
781 - C a.a. 32-45{B8, destruction boxa-catenin binding region, a.a 118-146
mmmmmmmmmmmÊmmmmmmÊmmtmÊmÊfmmmm A P C binding-regiOn Axin-binding region , mmmmmmmmmmmmÊmmmmmmÊmmmmmmÊmmmmmÊmmmm Tcf/ LEF-binding TOgiOn Cadherin-binding region Transactivation function 32 33 34 35 36 37 38 39 40 41 42 43 44 45 D S G I H S G A T T T A P S
F-box recognition region
*necessary for ubiquitin-dependent degradation of the protein
Figure 1: (l-catenin protein, its interaction regions, and degradation box.
Bars in blue show the interaction regions of P-cateniu with certain other proteins as mdicated on figure.
Huber et al. determined the three dimensional structure of the central core
armadillo (arm) repeats of murine P-catenin (Huber et al. 1997). The arm repeats do not
form separable domains. By forming a compact unit, arm repeats are highly resistant to
proteolysis, and no protein with fewer than six arm repeats has been described so far
The canonical 42 a.a. arm repeat consists.of three a-helices, which are connected
by short loops re-orienting the polypeptide by 90°. There are some deviations in
canonical repeats. Helix 1 in repeat 7 is replaced by a loop. The other deviation is a 22
a.a. insertion in repeat 10. This insertion including Arginine at codon 550 provides the
repeat 10 with high flexibility (Huber et al. 1997). The first 10 arm repeats have positive
surface potential, most of which lies in a shallow grove. Huber et al. 1997 proposed that
acidic regions of cadherins, APC, and the Tcf (T-cell fector)-family transcription fectors
interact with the basic groove of p-catenin. (Protein Data Bank, Brookhaven, NY, with
accession codes 2bct and 3 bet.)
1.1.4 p-Catenin in Cell-Adhesion Complex
The majority of human tumors (80-90%) originate from epithelial cells. Normally
these cells are tightly interconnected (reviewed by Christofori and Semb 1999). Cell-cell
and cell-matrix interactions are mediated by trans-membrane cell adhesion receptors of
the cadherin and integrin families, respectively.
Catenins were discovered as proteins associated with C54oplasmic domain of
trans-membrane cadherin proteins (Cowin 1994). Cadherins are a multi-gene family of
trans-membrane glycoproteins that mediate Ca^^-dependent intercellular adhesion,
cytoskeletal anchoring and signaling (Takeichi etal. 1991). E-cadherin, a component of
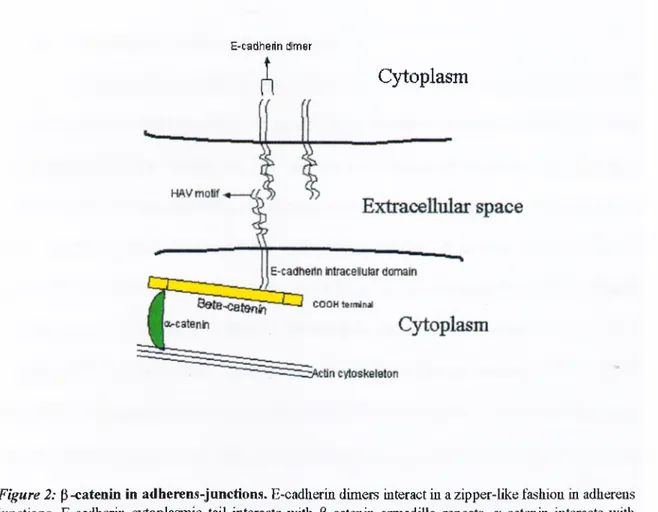
adherens junctions (reviewed by Yap et al. 1997), interacts with catenins [a, P, and y-
catenin (plakoglobin)], which link E-cadherin to actin cytoskeleton. Plakoglobin is a
homologue of P-catenin and also associated with adherens-type junctions, and in
et al. 1998). In adherens junctions, intracellular link between E-cadherin and actin
cytoskeleton is formed by catenins, which are a-catenin, P-catenin and y-catemn
(plakoglobin). P-catenin and y-catenin are members of armadillo protein family ( Peifer
et al. 1994). This protein family is characterized by a 42-aminoacid-repeat arm motif,
which was first found in the Drosophila segment polarity gene product armadillo
(Riggleman et al. 1989). P-catenin interacts with E-cadherin through its conserved central
region. The a-catenin, which interacts with amino terminal region of P-catenin, links P-
catenm to actin filaments (Figure 2).
E-cadherin dimer
Cjd:oplasm
Figure 2: p-catenin in adherens-junctions. E-cadherin dimers interact in a zipper-like fashion in adherens junctions. E-cadherin cytoplasmic tail interacts with P-cateuiu armadillo repeats, a-catenin interacts with the amino-terminal domain of P-catenin and with actiu cytoskeleton. In this way a and P-catenins coimect E-cadherin to actin cytoskeleton.
Tyr-654 phosphorylation of P-catenin is relevant for the modulation of in vivo
interaction between p-catenin and E-cadherin (Roura etal. 1999). Tyr-143 is the only Tyr
residue present in the P-catenin domain necessary for a-catenin binding (Barth et al.
1997). However the physiological relevance of tyrosine phosphorylation in the control of
p-catenin/a-catenin binding is likely to be dependent on the cell system (Roura et al.
1999).
Human cancer cells lines expressing a tmncated P-catenin show loose cell-cell
adhesiveness ( Kawanishi et al. 1995).
1.1.5 p-catenin as a bi-functional protein
Cadherin-based adhesive contacts are important for tissue organization in both
developing and adult organisms. As stated earlier, the trans-membrane protein E-cadherin
is involved in the linking of the plasma membranes of adjacent cells. Adhesion
complexes, in which cadherins participate, support calcium-dependent, homophilic cell
cell adhesion in all tissues of the body (reviewed by Christofori and Semb 1999).
Classical cadherins belong to a larger gene family of adhesion molecules including the
desmosomal cadherins, T-cadherin, Drosophila fat, the proto-oncogene ret, and the
others (Aberle et al 1996). E-cadherin extracellular segment consists of five cadherin
repeats. The cytoplasmic tail of E-cadherin binds to p-catenin, which in turn binds a-
catenin, and a-catenin is thought to be involved in interactions with the actin
cytoskeleton. Thus, P-catenin plays a major role in cell-cell interactions.
The second function of P-catenin was first delineated from homology between P-
discovery of a transcriptional role of ß-catenin indicated an unusual signal transducing
protein function of ß-catenin (reviewed Willert and Nusse 1998). The feet that mutations
of ß-catenin are found in colon cancer (Morin et al. 1997), melanoma (Rubinfeld et al.
1997), ovarian cancer(Palicios and Gamallo 1998; Sagae é ta l 1999), medullablastoma
(Zurawel et a l 1998), hepatocellular carcinoma (de La Coste et al. 1998), and some other
cancers is making this multifunction protein an attractive subject in cancer research.
1.1.6 Wnt Signaling
Wnt pathway of vertebrate is the homologue of Drosophila Wg (Wingless)
pathway. WntAVg signaling pathway is involved in a large variety of developmental
processes (reviewed by Cadigan and Nusse 1997). Wnt genes are defined by sequence
homology to the Wnt-1 gene in the mouse. To name, the functions of Wnt proteins are
embryonic induction, the generation of cell polarity, and the specification of cell fate in
development.
Wnts are secreted from cells rarely in a soluble form. The Porcupine (pore) may
be involved in secretion or ER transport. The pore gene product is a protein with eight
trans-membrane domains. Since embryos mutant for the segment polarity gene pore
(porcupine) show Wg mutant phenot}qje, pore might be required for Wg signaling
(Kadowaki et al. 1996).
To initiate Wnt signaling, Wnt protein interacts with its cognate Fz (Frizzled)
receptor (Bhanot et a l 1996). Several proteins bind to Wnts to modulate their activity.
Wnt and the frizzled receptor interactions may be regulated by glycosaminoglycans.
protein in a Drosophila cell line, and exogenous heparin can enhance Wg signaling
(Reichsman et al. 1996). Wg studies in Drosophila were the leading research area in
elucidating Wnt gene function in other organisms. In Wg signaling, embryos mutated in
porcupine (pore), dishevelled (dsh), armadillo (arm), pangolin (pan, D Tcj) have
segment polarity defects, however zeste-white (zw3), a homologue of mammalian GSK-
3P has opposite phenotype to those of Wg mutants. Fz relays WntA¥g signal through the
products of the dishevelled (dsh) and GSK-3p/zw3 genes (reviewed by Siegfried and
Perriman 1994).
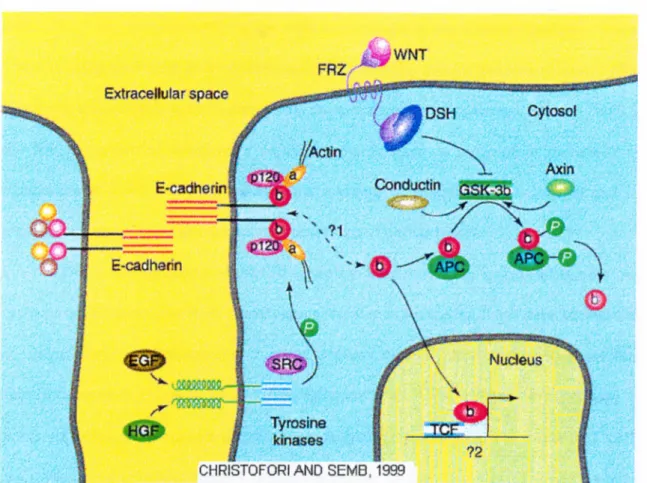
GSK-3p protein phosphorylâtes some serine-threonine residues on P-catenin. Wnt
signal stimulates the inactivation of GSK-3p kinase activity. APC (Adenomatous
polyposis coli) induces degradation of phosphorylated P-catenin (Rubinfeld etal. 1996).
Inactivation of GSK-3P by Wnt signal leads to accumulation of P-catenin in the
cytoplasm since a lack of phosphorylation of P-catenin precludes degradation by
ubiquitin/proteasome system (Miller and Moon, 1996). The APC protein appears to be a
negative regulator of cytoplasmic P-catenin (Korinek et al. 1997). The APC gene is the
most commonly mutated gene in colorectal cancer (85%). The early inactivation of APC
in colorectal cancer is associated with a rise in the free P-catenin. Colorectal tumors with
intact APC gene were found to contain activating mutations of p-catenin that altered
functionally significant phosphorylation sites (Morin et al. 1997). Accumulated P-
catenin/Arm enters the nucleus where it interacts with Tcf, alleviating its repression on
downstream genes. This interaction of Tcf and P-catenin modulates the expression of Tcf
3^'
Figure 3: A sum m ary of P-catenin functions. P-cateniu connects E-caciherin to actin cytoskcleton in adliereiis junctions (The function of P-catenin in cell-adhesion), fhe other role of P-catenin is in vvut signaling. Normally, p-catenin is phosphoiylaled by GSK-3P complex mid tliis tagged p-catenin is degraded by ubiquitin-proteasome pathway. In this way free P-catenin level is kept low. Wnt signal blocks GSK-3P activity, then unphosphoiylated free P-cateniu accumulates in the cytoplasm. Subsequently accumulated p-cateniu translocates into nucleus and binds to TCF/UZF-1 fmnily of transcription factors and releases the repression of transcription from down-stream genes. Wlien P-eatenin is mutated in putative phoshorylation sites in the mniuo-terminal domain, it becomes constitutively active. Axin iind conductin antagonize Wnt signaling by fonning a complex witli p-cateniu, AI^C, GSK-3P since this complex taigets P-catenin for degiadation. P-cateuiu is tyrosine phosphoiylated by the non-receptor tyrosine kinase SRC (Behrens et al. 1993). Tyrosine phosphorylation of P-calenin is a key step in the dissociation of P-catenin - E-cadherin (namely cytoskeleton-E-cadherin). Flazan and Norton showed that EGF could induce a dissociation of E-cadherin and P-catenin with vincuhn -which is a substitution of a-catenin-, a-actinin mid actin (Hazan and Norton 1998).
L1.7 Genes activated by Wnt signal
The Int-1 gene was initially identified as a gene involved in growth and
development signals in mouse mammary tumors (Nusse 1982, van Ooyen 1984). The
name int-1 was given since this gene became activated when Mouse Mammary Tumor
Virus is integrated next to it on chromosome 15 in the mouse genome (Nusse 1984).
However, a role of this gene in embryonic development was discovered, later on. Int-1 is
the mouse version of wingless, a segment polarity gene in Drosophila (reviewed by
Klingensmith 1994). Wnt-1, which is a melding of wingless (Wg) and Int and its
relatives are involved in developmental processes (Pennisi 1998).
The over-expression of c-MYC gene has been known in colorectal tumors. An
explanation for this came from chromosome transfer experiments. It has been shown that
an extra copy of chromosome 5 could repress c-MYC transcription and inhibits
neoplastic growth (Goyette et al 1992; Behrens et al. 1996/ When Wnt pathway is
activated, C54osolic ß-catenin accumulates, migrates into nucleus, and binds to T cell-
factor/lymphoid-enhancer-factor (Tcfi'Lef) proteins. In this complex, Tcf/Lef proteins
function as a DNA binding domain, and ß-catenin as a potential trans-activation domain
(van de Wetering et al. 1997). He et al. showed that expression of c-MYC is repressed by
wild-type APC and activated by ß-catenin. The results of this experiment fit with the
chromosome 5 transfer experiments since APC gene is located on chromosome 5q21.
According to data presented by He et al., the region of the c-MYC promoter that
conferred ß-catenin responsiveness was the same region shown to be APC-repressible in
colorectal cancer cells.
Matrilysin is a matrix metalloproteinase expressed in the malignant epithelia of
approximately 90% of human colonic adenocarcinomas and in a minority of human colon
tumor cell lines (Newell et al. 1994). Crawford et al. showed that ß-catenin trans-
activates the matrilysin by abrogating Tcf-mediated repression of matrilysin. Matrilysin
promoter contains an optimal Tcf-4 site downstream of the transcriptional start site that
interacts with P-catenin in vivo (Crawford etal. 1999).
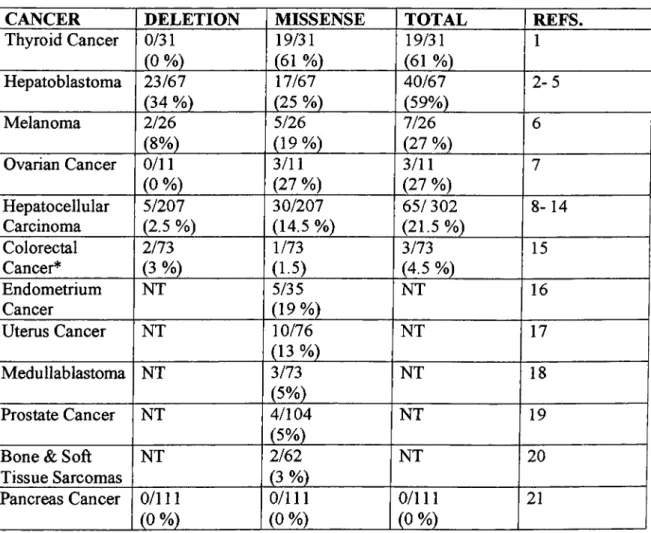
1.2.1 Mutations of p-Catenin gene
Mutations in the y^catenin regulatory domain and APC are mutually exclusive
The mutations in p-catenin generally affect exon 3.
Table 1: Somatic mutations of P-catenin in human cancers
CANCER DELETION MISSENSE TOTAL REFS.
Thyroid Cancer 0/31 (0 %) 19/31 (61 %) 19/31 (61 %) 1 Hepatoblastoma 23/67 (34 %) 17/67 (25 %) 40/67 (59%) 2-5 Melanoma 2/26 (8%) 5/26 (19%) 7/26 (27 %) 6 Ovarian Cancer 0/11 (0 %) 3/11 (27 %) 3/11 (27 %) 7 Hepatocellular Carcinoma 5/207 (2.5 %) 30/207 (14.5 %) 65/302 (21.5%) 8-14 Colorectal Cancer* 2/73 (3 %) 1/73 (1.5) 3/73 (4.5 %) 15 Endometrium Cancer NT 5/35 (19%) NT 16 Uterus Cancer NT 10/76 (13 %) NT 17 Medullablastoma NT 3/73 (5%) NT 18 Prostate Cancer NT 4/104 (5%) NT 19
Bone & Soft Tissue Sarcomas NT 2/62 (3 %) NT 20 Pancreas Cancer 0/111 (0 %) 0/111 (0 %) 0/111 (0 %) 21
* Colorectal cancers display APC mutations at high frequency (85 %) with functional activation o f P- catenin. References: 1) Garica-Rostan et al., 1999; 2) Koch et al. 1999; 3) Balker et al, 1999; 4) Jeng et al., 2000; 5) Wei et al, 2000; 6) Rubinfeld et al., 1997; 7) Palacios AGamallo 1998; 8) Miyoshi et al., 1998; 9) Satoh et al., 2000; 10) de la Coste et al., 1998; 11) Nhieu et al., 1999; 12) Huang et al., 1999; 13) Kondo et al., 1999; 14) Renard et al. 2000; 15) Sparks et al., 1998; 16) Kobayashi et al., 1999; 17) Fukuchi et al.,
1998; 18)Zuraweletal, 1998; 19) VoeDer et al., 1998; 20) Iwao et al., 1999; 21) Gerdes etal., 1999.
Table 2: Mutation spectra of P-catenin gene in human cancers*
CODON WT MUT MUT MUT MUT MUT
32 GAC TAC AAC GGC GTC GCC
D Y N G V A
33 TCT TGT T IT CCT CTT
S C F P L
34 GGA GTA GAA AGA
G
V E R 35 ATC AGC I S 36 CAT CCT TAT H P Y 37 T C T ^ TAT GCT TTT TGT S Y A F C 38 g g tI GAT^ G D 39 GCC A 40 ACT A IT T I 41 ACC GCC A lC T A I 42 ACA A lA T I 43 GCT GTT A V 44 CCT TCT Ps
45 TCT T IT CCT TAT TGT s F P Y C 46 CTG GTG L V 47 AGT AAT S N 48 GGT G 49 AAA AGA K R 50 GGC GAC G D*See table 1 for references.
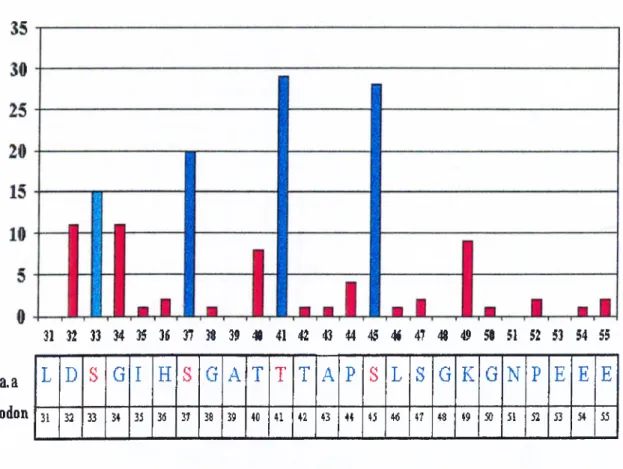
As shown in table 2, missense mutations occur at certain hot spots, which are
32D, 33S, 34G, 37S, 41T and 45S. However, anaplastic thyroid carcinoma has also
another hot spot, 49K (Lysine). This amino acid changes only to R (Arginine). Thyroid
carcinomas display also some other mutations; A20V, V22A, P52L, E54K, E55K, D58N
and S60F not reported in other tumors (Garcia-Rostan et al. 1999).
31 32 33 34 }5 36 37 J8 39 « 41 42 43 44 45 4i 47 « 49 58 51 52 53 54 55
L D S G I H S G A T T T A P S L S G K G N P E E E
31 32 33 34 35 3(5 37 38 39 40 41 42 43 44 45 4(5 47 48 49 50 51 52 53 54 55 a.a
codon
Figure 4: Hot spots for m utations in P-catenin. Putative phosphorylation sites 33S, 37S, 41T, and 45S are colored blue. Numbers at left side indicate the number of mutations reported so far. See table I for references.
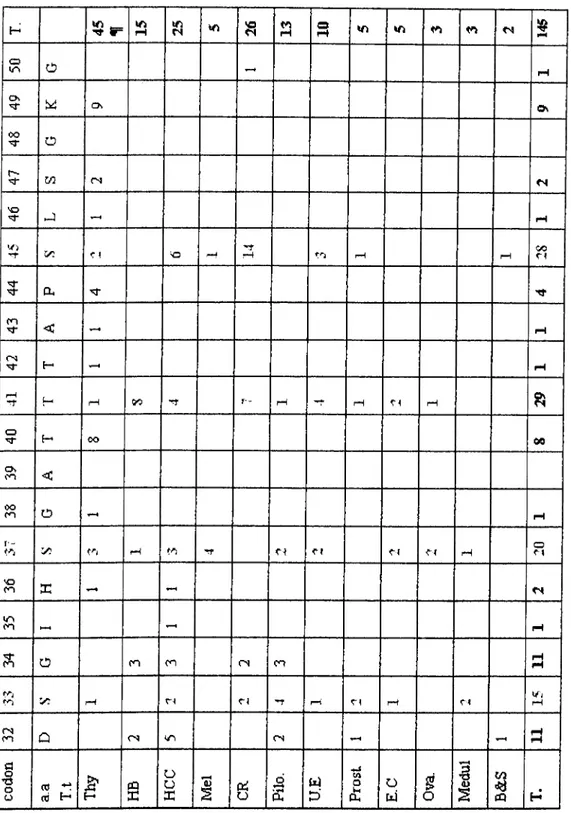
Tabu 3:Number of the mutations detected in [}-catenin (between a.a. 32 and 50 of exon 3). IM; tumor type. Thy; anaplastic thyroid carcinoma, ITB; Hepatoblastoma, ITCC; heptatocelhilai· carcinoma, Mel; melanoma cell lines, Pilo; pilomatricoma, CR; colorectal cancer, U.E; uterine endometrium cancer, Prost; prostate cancer, E.C; endometroial cancer. Ova; ovarian cancer, Medul; medullablastoma, B&S; bone and sol\-tissue tumors, T; Total. (^[, Mutations at codons 20, 22, 52, 54,55, and 60 are not included).
H Tf t= pH in VO iH orH cn cn o o rH Os Os 0\ oo O m (N <N V—1 rH rf ■y: c < rH H-rH rH rH GOCl Pц H* on < 1— 1 rH CN H rH rH rH OO r— rH rH pH ?; o OO 00 Os < oo on o rH r - y? rH cn H· CN rH oM 'O cn X ♦—· CN ITS on H-4 iH cn o cn cn CN cn rHiH y: rH H rH CN rH c< rH rs cn Q kTi ON iHpH § T5o o OJod H H cQX 1 O o (D 2 ceio ffi Dbl ■Hcn o H cu Ofri o <u H 15
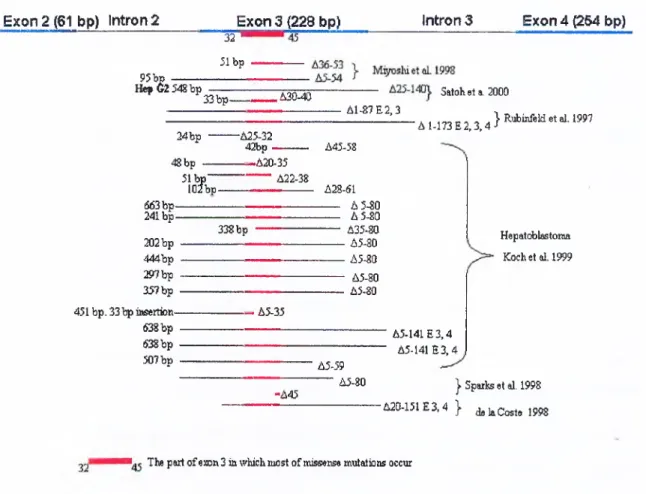
As shown in f ig u r e 5 , deletion type mutations in P-catenin gene are generally in
frame, and occur around exon 3, especially deleting conserved phosphorylation sites. As
a result, even though deletions occur in the N-terminal regions of the gene, the rest of the
protein is expressed since frames are conserved.
E xo n 2 (61 bp) in tro n 2 E x o n 3 (2 2 8 bp) Intron 3 E xo n 4 ^ 6 4 bp) 3 2 " 31 bp 95bp ---H ep62 54Sbp 33 b p - ,A30-40 Satoheta. 2000 A1-87E2,3 24bp -A25-32 42bp -'A 1-173 E 2 ,3,4}Rubinfeld etal. 1997 48bp — 51 b p “" lOfbp -A20-35 --- A22-38 663 b p - 241 b p -202 bp 444bp :S»7bp 357bp 451 bp. 33 Ip ittsertion- 638 bp 638bp . 507 bp . 338 bp A45-58 A28-61 --- A 5-80 --- A 5-80 --- A35-80 ---A5-80 --- A5-80 A5-80 A5-80 Hepatoblastoma Kochet al. 1999 . A5-35 A 5441E 3,4 A5-141E3,4 A5-59 --- A5-80
-A45 I Sparlss et al. 1998 -A20-151E3.4 } d.laC ost. 1998 The part of exon 3 in which most of missense mutations occur
Figure 5: Deletions in p-catenin gene and the deleted amino acids in the protein. The bp represents the deleted base pah's in the gene. Residues 32 to 45 are the part o f P-catenin in which most of missen.se mutations occur. A incicates deleted amino acids in the protein.
Gerdes et al. examined pancreatic tumors for P-catenin exon 3 mutations. They
have not found mutations in exon 3 of the p-catenin gene in neither the 111 exocrine nor
endocrine pancreatic tumors nor the 14 pancreatic cancer cell lines. They did not identify
intracellular P-catenin accumulation in any of the 40 pancreatic adenocarcinomas
examined (Gerdes et al. 1999).
Harada et al. constructed mutant mice expressing stable P-catenin, in which exon
3 deleted, in the intestine. These mice developed numerous intestinal adenomatous
polyps and some microadenomas (Harada et al. 1999)
1.2.2 Mutations of P-catenin in experimentally induced animal tumors
In contrast to human colon tumors, only 15 to 59 % of rat colon tumors induced
by heterocyclic amine dietary carcinogens have mutations in the Ape gene (Kakiuchi et
al. 1995), but all others without an APC mutation had mutations in the p-catenin gene
(Dashwood et al. 1998). Suzui et al. studied p-catenin mutations in
Methyazoxymethanol (MAM) acetate induced rat colon adenomas and adenocarcinomas.
They screened genomic DNA samples including exon 2, intron 2 and exon 3, which
showed 100% homology to mouse P-catenin, and codons 33,37,40,41,42, and 45 code
for serine or threonine residues. In this study, Suzui et al. found three types of mutations
in 75% of adenomas and 94% of adenocarcinomas. Of 18 mutations out of 20 tumor
samples, 72% were G to A, 17% were to A, and 11% were C to T mutations.
Of these only to T (Thr 41 lie) substitution affected a possible phosphorylation site
(Suzui e ta l.l 999).
Ogawa et al. used B6C3F1 mice to induce hepatic adenomas and HCC by
Diethylnitrosamine (DEN). All mutations were in codon 41 of p-catenin (in exon 3),
which is a possible phosphorylation site for GSK-3p. They detected transitions from
ACC (threonine) to АТС (isoleucine) in 5 of 13 HCC and from ACC (threonine) to GCC
(alanine) in 2 of 13 HCC. However, they did not identify any mutations in any of 14
adenomas. This implied that p-catenin mutation is a late event during hepatic
carcinogenesis. When compared to other studies where p-catenin mutations are
distributed on and/or around possible phosphorylation sites, this study resulted in
mutation only at codon 41. This may imply that different chemicals induce mutations by
different mechanism (Ogawa et al. 1999)
Tsujiuchi et al. induced tumors in male Fischer 344 rats with N-
nitrosodiethylamine and a choline-deficient L-amino acid-defined diet. They then
extracted total RNA from individual lesions and investigated mutations in the GSK-Зр
phosphorylation consensus motif of P-catenin by reverse transcriptase-PCR-single-strand
conformation polymorphism analysis followed by nucleotide sequencing. Changes were
detected in 5 of 11 HCCs induced by the exogenous carcinogen. The observed changes
C: G to G: C or C: G to A; T at codon 33 and G: C to T; A transversions at codon 34.
These mutations were associated with p-catenin protein accumulation and confirmed by
Western blot analysis. In contrast to chemically induced HCCs, only 2 of 15 HCCs
induced by choline-deficient diet displayed mutations, those being transitions of C:G to
T:A at codon 41 without amino acid alteration (Tsujiuchi etal. 1999).
1.2.3 The accumulation of p-catenin protein in tumor cells
The accumulation of P-catenin has been observed in tumors with or without P-
catenin mutations. The APC and Axin protein is involved in the active degradation of p-
catenin. Some tumors with APC and/or Axin mutations show abnormal accumulation of
P-catenin in nucleus or abnormal distribution in cytoplasm. Normally, P-catenin is
observed along the cellular membrane.
Anaplastic thyroid carcinoma samples display nuclear accumulation in 42% of the
samples. The high accumulation of the P-catenin in nucleus correlated with the
aggressiveness of this tumor. In the study of Garcia-Rostan et al. 1999, all of the patients
died within 1 year of diagnosis. (Garcia-Rostan et al. 1999)
In hepatoblastomas (HB), p-catenin strongly accumulated in the cytoplasm and
nucleus as shown by immunohistochemical staining. However, in normal liver P-catenin
is localized along the cytoplasmic membrane (Koch et a/. 1999). In another study on
hepatoblastomas, nuclear and cytoplasmic accumulations were demonstrated in 100% of
samples, even though P-catenin gene was mutated in only 34% of the samples (Blaker et
al. 1999).
In HCC, the accumulation of p-catenin was resulted from its own mutations or
Axin mutations. APC is rarely mutated in HCC. SNU449 and HepG2 cell lines (both
have mutated P-catenin) display strong nuclear staining. Alexander (PLC), SNU423, and
SNU475 cells display a spotty cytoplasmic and a moderate nuclear staining for p-catenin.
These three cell lines ha-we AXIN 1 mutations (Satoh et al. 2000).
Rubinfeld et al. showed defective P-catenin regulation in 8 of 26 melanoma cell
lines because of P-catenin mutations, unusual mRNA splicing, or inactivation of APC
(Rubinfeld ei a/. 1997).
In colorectal cancers, APC is mutated in 80% of all cases. However, P-catenin
mutations are found in 50% of the cases without APC mutations. Shimizu et al. showed
that 84.4% of carcinoma in adenomas (CIA) displays the nuclear localization of P-catenin
(Shimizu et al. 1999)
Uterine endometrium has P-catenin mutations (13%). However 30% of the
samples without P-catenin display P-catenin accumulation in cytoplasm and/or nucleus.
While, 90% of samples with P-catenin mutations display P-catenin accumulation
(Fukuchi etal. 1998).
In endometrial carcinomas, 3 of 17 cases display P-catenin accumulation in the
nucleus and cytoplasm, one of the tumors without any p-catenin mutation (Kobayashi et
a/. 1999).
63% of bone and soft-tissue tumors without any p-catenin mutations in exon 3
dislpay P-catenin accumulation (Iwao et al. 1999).
An APC truncation mutation was found to associate with elevated cytosolic P-
catenin level in a breast cancer cell line (Schlosshauer et al. 2000)
In conclusion, mutations affecting P-catenin, APC or Axin (Dceda et al. 1998)
gene result in the accumulation of P-catenin protein in tumor cells. This accumulation is
reflected by an increase in the cytoplasmic and nuclear P-catenin levels.
1.3. Tumor Antigens, Tumor Associated-Antigens and Autoantibodies
1.3.1 Tumor Antigens
Tumors are derived from self-tissues in nature, however malignancy may be
associated with the expression of molecules on cells, which are recognized as foreign by
the immune system. These molecules are called “tumor antigens”. Theoretically, immune
system could recognize and destroy these tumor antigen-expressing cells before they
grow into tumors. This hypothetical function is called “immunosurveillance”. This term
correlates with the feet that immunodeficient individuals are more likely to develop
certain types of cancers compared to normal individuals. However, tumor cells are able
to escape the immunosurveillance, as lethal cancers arise in immunocompetent
individuals. Tumor immunology is a common field of immunology and oncology
studying the specific acquired immunoresponses, the tumor or tumor-associated antigens,
effector mechanisms, diagnosis, and finally treatment of tumors.
Tumor antigens may be classified in two groups. First group is tumors antigens
recognized by T lymphoc3^es. This type of tumor antigens are cell proteins, which have
been processed and presented to CD4+ or CD8+ T cells as antigen-MHC complexes.
Second type of tumor antigens are identified by an antibody response in tumor bearing
animals or humans (reviewed by Sinkovics, Horvath, 2000). The p53 protein has been
initially identified as a tumor antigen, in animals carrying of this type of SV40-induced
tumors (Lane and Crawford, 1979).
The altered forms of tumor suppressor and proto-oncogene products expressed in
tumors are generally intracellular molecules which are likely to be processed and
presented as peptides associated with MHC I, however phagocytosis or internalization of
tumor cells may result in MHC П-associated presentation (see Soussi T, 2000).
1.3.2 Tumor-Associated Antigens (TAA)
Most of tumor antigens are shared by different tumors arising from the same types
of cells, and most may also be found on some normal or benign tumor cells. It is
suspected that many of these molecules may not stimulate immunoresponse in tumor
host. Such antigens are often called tumor-associated antigens (reviewed by Roselli et al.
1996) identified by polyclonal or monoclonal antibodies generated against tumor cells or
purified proteins. This type of antigens may be important for diagnosis. The most known
characterized TAAs are alpha-fetoprotein (AFP) and carcinoembryonic antigen (CEA).
AFP is normally expressed and secreted in fetal life by yolk sac and liver, but in
adult life the protein is replaced by albumin and is present at low levels. Serum levels of
AFP may be significantly elevated in patients with hepatocellular carcinoma or germ cell
tumors. Elevated AFP in sera may be used as indicator of liver or germ cell tumors or of
recurrence of these tumors afl:er treatment. The limitation is that elevated serum levels
are also found in non-neoplastic liver diseases, e.g. cirrhosis (reviewed by Johnson 1999).
1.3.3 Autoantibodies in Tumor Hosts
The tumor suppressor protein p53 is hardly detectable in normal cells (Crawford
et a/. 1983). TP53 gene encoding the p53 protein is the most frequently mutated tumor
suppressor gene in human cancers (reviewed by Hoolstein et al. 1991). When p53 is
mutated in cancer cells, it is not able to control cell proliferation resulting in genetically
unstable cells. The most of the changes in p53 genes are missense mutations and occur
generally at DNA-binding domain (Hollstein 1991). More than 90 % of the point
mutations occur between exon 4 and exon 10 within DNA binding domain (Beroud and
Soussi, 1998). Inactive mutant p53 protein stability is increased to several hours of half-
life from 20 minutes of half-life of normal p53 and accumulates in the nucleus (Casey et
al. 1996).
Crawford et al. first described autoantibodies in breast cancer sera (Crawford et
al. 1982). These studies were ignored for ten years and prompted after the discovery that
p53 is altered in almost every type of cancer. There is generally a significant correlation
between auto-p53 antibodies and p53 accumulation and/or mutations. So fer, it has been
shown that 20- 40 % of patients with p53 mutations have anti-p53 autoantibodies
(reviewed by Soussi, 2000). However, some patients with wild type- (or undetectable
mutations in) p53 also develop p53 autoantibodies. It was shown that p53 autoantibodies
strongly bind the amino and to a lesser extent, carboxyl-terminus of the protein, although
the missense mutations do not generally occur in these regions (SchlichthoLz et al. 1992).
This indicates that p53 autoantibodies can recognize both mutated and wild type-p53
protein. Also, the immunizing of mice with human wild type p53 triggers the production
of antibody formation against linear epitopes in the amino and carboxy termini (Legros et
al. 1994). The p53 autoantibodies are generally IgGl and IgG2 subtypes. Some patients
have predominantly IgA response (Lubin et al. 1995). No patient has only IgM response
and IgG3 or IgG4 responses have not been detected (reviewed by Soussi 2000). Detection
kits of p53 antibodies in sera by using ELISA have been developed. Mutant p53
accumulation seems to be an important factor in this humoral response in addition the
genetic background may be important in developing anti-p53 autoantibodies.
Zalcman et al. monitored the p53 autoantibodies in lung cancer patients by using
ELISA for 30 moths during the treatment. They reported that 12 of 16 antibody positive
patients had reduced titers during chemotherapy that led to partial or complete remissions
of disease. They commented that the very rapid, specific decrease in these antibodies
during therapy suggests that a constant level of tumoral cells with increased p53 protein
is necessary for a detectable humoral anti-p53 response (Zalcman et al. 1995)
Hallak et al. showed that 18% of patients suffering from colorectal cancer
developed autoantibodies against p53 in their sera, which might be an early indicator for
tumor development and distant metastasis (Hallak et al. 1998^
Maass et al. screened sera from Squamous Cell Carcinoma of Head and Neck
(SSHN) and concluded that the frequency of p53 antibodies in these patients is
comparable with the occurrence of these antibodies in other malignancies (Maass et al.
1996).
In lung cancer, p53 autoantibodies are high (30%) and are correlated with a very
high rate of p53 mutations in this cancer (60-70%). The autoantibodies are usually IgG,
which means that a secondary response occurred after a prolonged immunization (Lubin
et al. 1995). Wild et al. demonstrated that serum antibodies were detected only in a
proportion of lung cancer cases, 15% of patients with p53 mutation. They concluded that
the majority of serum p53 antibodies were specifically associated with a detectable p53
mutation in the tumor (Wild et al. 1995). Green et al. concluded from the study of the
expression of the p53 gene and presence of p53 autoantibodies in ovarian cancer that
tissue expression of p53 in ovarian tumor is associated with poor histological
differentiation and the presence of detectable serum autoantibodies (Green et al. 1995).
p53 autoantibodies have been also found in other body fluids. Abendstein et al.
demonstrated serum and ascitic anti-p53 antibodies in 25% and 19% of the ovarian
cancer patients studied respectively and concluded that the generation of a humoral
immune response against p53 protein in the close tumor environment is associated with
poor disease free survival (Abendstein et al 2000). Such antibodies have be also found
saliva of oral cancer patients (Tavassoli et al. 1998) and the pleural effusion of
pancreatic, colon and lung cancer patients (Munker et al. 1996)
Antibodies against a few oncogenes have been described. Sorokine et al. found
that human sera contain IgG antibodies to c-myb gene product in cancer patients
(Sorokine et al. 1991). They concluded that the percentage of positive sera in cancer
patients appears to be dependent on cancer type and is significantly higher in breast
cancer patients (43%) than in normal donors.
Ben-Mahrez et al. found c-myc IgG autoantibodies in 25 of 44 sera fi’om
colorectal cancer patients. However, they also found autoantibodies in 8 of 46 sera fi'om
normal donors (Ben-Mahrez et al. 1990).
In another study, Yamamoto et al. found autoantibodies against c-Myc in 9 of 68
(13.2 %) lung cancer patients and 1 of 30 (3.3%) healthy volunteers without any
circulating c-Myc antigen in either group. These antibodies were against exon 2 alone
(4/68), exon 3 alone (1/68) or both exon 2 and exon 3 (4/68) of c-Myc (Yamamoto et al.
1999).
Covini et al. demonstrated autoantibodies against cyclin B1 in sera from 15 of
100 patients with HCC. They found only one serum reactive against to cyclin A and
another one reactive with CDK2 but no sera reactive with cyclin D1 and E (Covini et al.
1997).
1.3.4 Cytotoxic T cell (CTL) responses against p53 and P-Catenin
Normally, in body endogenous human CTL response to p53 is weak (McCarty et
al. 1998). A hot spot mutation at a.a. 273 from R to H in p53 protein renders cells
resistant to killing by HLA-restricted c5l:otoxic T lymphocytes (CTLs) specific for
epitope 264-272 a.a. The R to H mutation alters proteasomal processing of the p53
protein by inhibiting cleavage between a.a. 272 and 273. This prevents the release of the
CTL epitope spanning residues 264-272 (Theobald et al. 1998).
Ciernik et al. transfected the human cell line Hmy-2.C1R and the p53-null human
lung cancer cell lines H358 and H1299 with an expression vector encoding a mutant-
human p53 (Cysl35Tyr). Then cells were tested as targets for murine mutation-specific
CTLs. They demonstrated that these human lung cancer cells effectively process and
present this endogenous mutant p53 epitope resulting in mutant epitope-specific lysis by
CTLs (Ciernik et al. 1996).
Peralta et al. studied whether human bladder cancer xenografts in SCID mice
could be eliminated by CTLs, which recognize over-expressed p53. They concluded that
overall response of the human bladder tumors in the SCID mouse model suggests the
possibility of targeting p53 in patients with bladder cancer (Peralta et al. 1999).
Barfoed et al. generated a number of CTL clones recognizing the HLA-A*0201
p53 wt peptide RMPEAAPPV (65-73) by the in vitro stimulation of CD8 enriched PBL
from a healthy HLA-A*0201 donor using peptide loaded autologous dentric cells
(Barfoed et al. 2000).
Robbins et al. reported that the mutated product of the P-catenin gene was
recognized by melanoma-specific tumor infiltrating T cells (TEL 1290). The mutation
appeared to convert a peptide from a very low affinity binder to a very high affinity
binder of the HLA-A24 class I gene product. A peptide corresponding to the sequence
between a.a. 29 and 37 of the mutant gene product (Ser37Phe) was identified as T cell
epitope recognized by TIL 1290 (Robbins et al. 1996).
The comparative study of P-catenin and p53 in different human tumors allowed
us to ask the following question: can cancer patients develop autoantibodies to P-catenin
protein as a result of its accumulation in certain type of tumor cells, similar to p53
autoantibodies? Thus, we decided to set up an experimental system to screen cancer sera
for possible anti- P-catenin autoantibodies.
For this aim, we first needed purified P-catenin antigen. We aimed to obtain such
an antigen as a recombinant protein in E. coli. We decided to subclone a P-catenin cDNA
fragment into a bacterial expression system. For technical reasons, we planned to obtain
an N-terminally deleted p-catenin protein (lacking first 88 a.a. residues). As many cancer
cells display an N-terminally deleted p-catenin (AN-p-catenin) protein product, we
assumed that, such truncated proteins would also be immunogenic in cancer patients. In
addition, as stated earlier, p53 autoantibodies are directed to both N-terminal and C-
terminal regions despite the feet that most p53 mutations affect the central domain.
In order to fecilitate the screening of a large number of cancer sera, an ELISA-
based test was chosen as a first screening method. We reasoned that, once we identified a
small number of candidate cancer sera for anti P-catenin autoantibodies, we could
perform additional detection methods such as western blotting and immunoprécipitation. 1.4 AIM AND STRATEGY
2.1 MATERIALS
2.1.1 Chemicals
The laboratory chemicals were analytical grade and supplied from Sigma
Biosciences Company Limited (St. Louis, MO, U.S.A) or from Carlo-Erba (Milano,
Italy)
2.1.2 Bacterial Strains
Escherichia coli (E.coli), DHSa: F’, (f80dE(lac Z)M15), rec A l, endAl,
gyrA96, thil, hsdR17, (r-km-k), supE44, relAl, deoR, E(lacZYA-ar gF)U169
M15: Another E.co// strain and with pREP4 plasmid.
JM109: E.coli strain.
2.1.3 Enzymes
Restriction endonucleases (Sac I and Sal I) and T4 DNA Ligase were purchased
from MBI FERMENTAS Inc. (NY, U.S.A)
2.1.4 Electrophoresis
Electrophoresis grade agarose was supplied from Sigma (St. Louis, MO, U.S.A).
2.1.5 Antibodies
The monoclonal antibody (6B10) against p53 was obtained from within the
department (Yolcu etal., unpublished results).
The monoclonal antibody P-catenin (E-5) was purchased from Santa Cruz
Biotechnology, Inc. (U.S.A) This antibody was generated against carbox)4erminal region CHAPTER 2: MATERIAL AND METHODS
(a.a. 680-781) of human P-catenin and it does not cross-react with N-catenin, alpha E-
catenin or gamma catenin.
Secondary antibodies (Anti mouse-HRP/-AP, Antihuman-HRP/-AP) were
supplied from Dako.
2.1.6 Kits
QIAGEN Plasmid purification kit (by Qiagen, Chatsworth, CA, U.S.A) was
utilized to obtain plasmid DNA at high amounts and concentration with high purity.
TNT Coupled Reticulocyte Lysate System was purchased from Promega
(Madison, WI, USA) for the single step in vitro transcription-translation (IVTT) of genes
in a eukaryotic system.
Enhanced Chemiluminescence (ECL) system from Amarsham (Uppsala, Sweden)
provided the means to visualize the HRP conjugated antibodies in Western Blotting.
Amplify from Amersham (Uppsala, Sweden) is a fluorimetric method to enhance
the visualization of the signals generated by radioisotopes on polyacrylamide gels.
2.1.7 DNA and Protein Size Markers
DNA size marker, 1 kb DNA ladder was from MBI FERMENTAS Inc. (NY,
U.S.A).
SDS-PAGE protein size markers were from either Pharmacia (Uppsala, Sweden)
or from Sigma (Saint Louis, Misspuri, U.S.A), (rainbow marker).
2.2 SOLUTIONS AND MEDIA 2.2.1 Agarose Gel Electrophoresis (AGE) Solutions
Tris-acetic acid-EDTA (TAE): 50 X stock solution: 242 g Tris base, 57.1 ml Glacial
acetic acid, 100 ml of 0.5 M EDTA are dissolved in 1 liter water. IX working solutions
includes 40 mM Tris-acetate and ImM EDTA.
Ethidium bromide: The stock solution: 10 mg/ml in water. Working solution: 30 ng/ml.
2.2.2 Solutions for Plasmid DNA isolation (mini-prep)
Solution /: 50 mM Glucose, 25 mM Tris.Cl pH 8.0,10 M EDTA, should be autoclaved. Solution II: 0.2 N NaOH, 1 % (w/v) SDS
Solution III: 3 M Potassium acetate pH 4.8,11.5 % (v/v) glacial acetic acid. Phenol/Chloroform: 100 % equilibrated phenol/ 100 % chloroform.
2.2.3 Solutions for bacterial transformation
CaCE: 50 mM in double distilled water and filter sterilized.
Transformation Buffer (ТВ): 10 mM K.PIPES, 55 mM MnCb, 15 mM CaCb, 250 mM
KCl. Filter sterilized and stored at 4 °C.
2.2.4 Microbiological media and antibiotics
Luria-Bertani Medium (LB): 10 g tryptone, 5 g yeast extract, 5 g NaCl and 1 ml of 1 N
NaOH are dissolved in 1 liter water. For LB-plates, 15 g/L bacto agar is added. LB is
sterilized by autoclaving.
SOB medium: For 1 It., 20 g tryptone (2%), 5 g yeast extract (0.5%), 0.584 gr NaCl (10
mM), 0.1864 g KCl (2.5 mM) autotclaved to sterilize. Then, 2.46 g MgS0 4and 2.03 g
MgCb (10 mM) are added.
SOC medium: SOB + 20 mM glucose from filter sterilized 1 M glucose stock solution in
ddHzO.
Ampicillin: Stock solution, 100 mg/ml in ddH20 and stored at -20 "C. Working solution,
100 |xg/ml.
Kanamycin: Stock solution; 25 mg/ml solution in ddH20 and stored a t-20 °C. Working
solution; 25 |J.g/ml.
0.1 M IPTG: 1.41 g IPTG (isopropyl-p-D-thiogalactopyronoside); water is added to 50
ml, sterilized by filtering.
2.2.5 Polyacrylamide Gel Electrophoresis Solutions
SDS-PAGE was prepared at 8 or 10% concentrations for resolving gel, and 5% for
stacking gel.
Running Buffer (Tris-glycine electrophoresis buffer): 25 mM Tris base, 250 mM glycine.
1%SDS.
Sample dye (cracking buffer) (2x): 50 mM Tris.HCl pH 6.8,1% SDS, 2 mM EDTA, 1%
2-ME, 0.02% Bromophenol Blue, 10% glycerol.
30% acrylamide-bisacrylamide solution: 29 g of acrylamide and 1 g of bisacrylamide is
dissolved in 100 ml ddH2 O. Stock solution is stored in dark at 4 °C.
10% SDS: A 10% (w/v) stock solution is prepared in deionized water and kept at room
temperature.
10%APS: A small amount (such as 0.1 g) needed is weighed and dissolved in de-ionized
water at final 10% and stored in dark and 4°C for at most one week.
Coomassie Staining Solution: 0.25 g of Coomassie Brilliant Blue R250 is dissoleved in
90 ml of methanol: ddH2 O (1:1 v/v) and 10 ml of glacial acetic acid.