T.C.
EGE ÜNİVERSİTESİ TIP FAKÜLTESİ
ÇOCUK SAĞLIĞI VE HASTALIKLARI ANABİLİM DALI PROF. DR. SAVAŞ KANSOY
ÇOCUKLUK ÇAĞI SANTRAL SİNİR SİSTEMİ TÜMÖRLERİNİN BİR ALT GRUBU OLAN
EMBRİYONEL TÜMÖRLÜ OLGULARDA HSNF5/INI1 GEN MUTASYONLARININ ARAŞTIRILMASI UZMANLIK TEZİ DR. ELİF KIYMET Tez Danışmanı
PROF. DR.FERDA ÖZKINAY
ÖNSÖZ VE TEŞEKKÜR
Çocuk hekimi olma yolculuğumda, bilgi ve deneyimlerini benimle paylaşarak eğitimime katkıda bulunan, kendimi bu büyük ailenin bir parçası gibi hissetmemi sağlayan başta Çocuk Sağlığı ve Hastalıkları Anabilim Dalı Başkanımız Sayın Prof. Dr. Savaş KANSOYolmak üzere tüm saygıdeğer hocalarıma ve uzmanlarıma,
Birlikte çalışma fırsatım olduğu için her zaman kendimi çok şanslı hissettiğim, yaptığı işe karşı duyduğu saygı ve heyecanı öğrencilerine aktararak onlara da Genetiği sevdiren, bilgi ve deneyimini tezimin her aşamasında benden esirgemeyerek yolumu aydınlatan, çalışma azmini her zaman kendime örnek alacağım saygıdeğer tez danışmanım Prof. Dr. Ferda ÖZKINAY’a
Tezimin oluşturulmasında, takıldığım her noktada yardımıma koşarak beni hiç yalnız bırakmayan sevgili Dr. Esra IŞIK ve Doç. Dr. Tahir ATİK’e, yine hiçbir yardım çağrımı geri çevirmeyerek bana bu süreçte destek olan Bilçağ AKGÜN’e,
Tezimin laboratuvar çalışmalarının yapıldığı Tıbbi Genetik laboratuvarında emek veren tüm çalışanlara,
Hasta seçimimde yardımcı olan Onkoloji Bilim Dalı Öğretim Üyelerinden değerli Prof. Dr. Nazan Çetingül’e ve Prof. Dr. Mehmet Kantar’a,
Hastalarımın doku örneklerine ulaşmamı sağlayan, kapısına her gidişimde beni hoşgörü ile karşılayarak yardımcı olan saygıdeğer Tıbbi Patoloji Bilim Dalı öğretim üyesi Prof. Dr. Taner AKALIN’a
Asistanlık süreci boyunca birlikte çalıştığım, ailemden çok vakit geçirdiğim ve ailem kadar çok sevdiğim tüm araştırma görevlisi arkadaşlarıma
Bu dört yıllık zorlu eğitim sürecinde beraber çalıştığım, karşılaştığım zorluklarla el ele vererek baş edebildiğim, geriye dönüp baktığımda “Dört yıl önce iyi ki bu kapıdan girmişim.” dedirten başta sevgili eş kıdemime ve tüm asistan arkadaşlarıma,
Doğduğum günden beri beni kendilerinden çok düşünen, arada mesafeler olsa da desteklerini nefesim kadar yakınımda hissetmemi
sağlayan, elde ettiğim her başarıyı onların alın terine ve sevgisine borçlu olduğum sevgili annem Sevcan KIYMET ve babam Mehmet KIYMET'e,
Beni benden çok düşündüklerini bildiğim ve benim de her zaman kendimden çok düşündüğüm, can yoldaşlarım, canım kardeşlerim Murat ve Melek KIYMET’e,
Sonsuz teşekkürlerimi sunarım.
Dr. Elif KIYMET Temmuz 2017, İZMİR
ÖZET
Amaç: Bu çalışmada, çocukluk çağı santral sinir sistemi tümörlerinin bir alt grubu olan embriyonel tümörlerden, atipik teratoid rabdoid tümör (AT/RT), tanısı almış olan hastalarda, bu tümör ile ilişkili olan bir tümör supresör gen (hSNF5/INI1) mutasyonunun araştırılması amaçlanmıştır.
Gereç ve Yöntem: 2010-2015 yılları arasında Ege Üniversitesi Tıp Fakültesi’nde AT/RT tanısı almış olguların dosya verileri incelendi. Klinik bulguları ve tümör dokusunda INI1 ekspresyonu açısından yapılmış olan immunohistokimyasal analiz sonuçları değerlendirildi. Olguların hepsinin tümör dokusuna ait patoloji preparatlarından ve ulaşılabilen olguların periferik kanlarından elde edilen DNA ile hSNF5/INI1 geni için tüm eksonik bölgeler PCR ile çoğaltılıp, jel elektroforezi ile kontrol edildikten sonra, DNA dizi analizi gerçekleştirildi. Elde edilen sonuçlar referans genom ile karşılaştırıldı. Tespit edilen varyasyonların patojenitesi ACMG 2015 kriterlerine göre değerlendirildi. Tümör dokusunda mutasyon tespitedilen hastalardan ulaşılabilenlerde periferik kan DNA’sında analiz tekrarlandı, ulaşılamayanlarda anne ve babada periferik kan DNA analizi yapıldı.
Bulgular:Çalışmamıza toplam 10 hasta aldık, bunların 7’si SSS’nin AT/RT’ü, 2’si yumuşak dokuda rabdoid tümör, biri renal rabdoid tümör tanılıydı. Santral sinir sisteminin(SSS) AT/RT’ü tanısı alan 7 hastanın 4’ü (%57) kız, 3’ü (%43) erkekti. Bu olguların ortanca tanı yaşı 18 ay (3 ay-12,9 yaş) bulundu. Hastalarımızın %85’i 36 aydan önce tanı almıştı. Hastalarımızın ortalama sağ kalım süresi 5,8 (±28,7)aydı.
SSS’nin AT/RT’ü tanısı ile izlenen olguların 4’ünde (%57) tümör infratentorial yerleşimli iken 3’ünde (%43) supratentorial yerleşimlidir. Supratentorial bölgeye yerleşen tümör nedeni ile izlenen hastalarda ortanca tanı yaşı 19 ay (16 ay-12,9 yaş), ortalama sağ kalım süresi 7,3 (±5,7) aydır. İnfratentorial bölgeye yerleşen tümör nedeni ile izlenen hastalarda ise
ortanca tanı yaşı 17,5 ay (3-19ay), ortalama sağ kalım süresi 23,2 (±38,5) aydır.
SSS’nin AT/RT’ü tanısı alan 7 hastadan 2’sine (%28,5) total eksizyon, 5’ine (%71,4) kısmi eksizyon yapılabilmişti. Total eksizyon yapılan hastaların ortalama sağ kalım süresi 43,5 (±53,7) ay bulunurken rezidü tümör dokusu kalan hastaların ortalama sağ kalım süresi 5,8 (±4,7) ay bulundu.
SSS, yumuşak doku ve renal rabdoid tümör tanılı hastaların tümör dokusunda immunohistokimyasal yöntemler ile hSNF5/INI1 protein ekspresyonu bakıldı ve %77 hastada protein ekspresyon kaybı bulundu.
Yumuşak doku ve renal rabdoid tümör tanılı hastaların hepsinde tümör dokusunda hSNF5/INI1 protein ekspresyon kaybı gösterildi.
SSS’nin AT/RT’ü tanılı 7 hastadan 4’ünde protein ekspresyon kaybı mevcuttu, bir hastada protein ekspresyonu tamamen korunmuştu, bir hastada ise protein ekspresyonu fokal olarak korunmuştu. Bir hastada protein ekspresyonu değerlendirilmemişti.
hSNF5/INI1 protein ekspresyon kaybı gösterilen hastaların ortanca tanı yaşı 18 ay (3-19 ay), ortalama sağ kalım süresi 3,2 (±2,6)aydı.
hSNF/INI1gen dizisinde 4 hastada, tümör dokusunda mutasyon saptandı. Bu hastaların 3’ünün SSS’nin AT/RT’ü tanısı, 1’inin yumuşak dokuda rabdoid tümör tanısı mevcuttu.
SSS’nin AT/RT’ü tanısı ile izlenen ve hSNF5/INI1 geninde mutasyon saptanan hastaların ortanca tanı yaşı 19 ay (17-19 ay), ortalama sağ kalım süresi 4 (±2,6) ay bulundu. hSNF5/INI1 geninde mutasyon saptanmayan hastaların ortanca tanı yaşı 17 ay (3 ay-12,9 yaş), ortalama sağ kalım süresi 25,7 (±37,1) ay bulundu.
Yumuşak dokuda rabdoid tümör tanısı ile izlenen ve tümör dokusunda hSNF5/INI1 geninde mutasyon saptanan bir hasta vardı. Bu hastanın tanı anında yaşı 3,5 ay, sağ kalım süresi 15 aydı. Bu çalışma yapıldığı sırada hastalığı remisyondaydı.
Tümör dokusunda mutasyon saptanan hastaların kanlarında elde edilen DNA’da mutasyon saptamadık.
Mutasyon taşıyan hastaların anne babaları aynı mutasyon açısından Sanger dizi analizi kullanılarak değerlendirildi. Hastaların anne ve babalarının bu mutasyonu taşımadıkları gösterildi.
Sonuç:AT/RT tanılı hastaların tümör dokusunda hSNF5/INI1 geninde mutasyon saptanması durumunda hastaların yaşam sürelerinin kısa olduğu gözlenmiştir. Bu nedenle prognozu belirlemede mutasyon analizi önerilebilir. Bu konuda daha geniş hasta gruplarında araştırma yapılmasına gereksinim vardır. Önceki yayınlardan farklı olarak AT/RT tanılı hasta grubumuzda kanda yapılan DNA analizlerinde hiçbir hastada hSNF5/INI1 geninde mutasyon saptanmamıştır. Bu sonucun kanda analiz yapılabilen hasta sayısının kısıtlılığı ile ilgili olduğu düşünülmüştür.
Anahtar kelimeler:Santral sinir sistemi tümörleri (SSS tümörleri), Atipik teratoid rabdoid tümör (AT/RT), hSNF5/INI1
SUMMARY
Objective: In this study, it was aimed to investigate the mutation of a tumor suppressor gene (hSNF5 / INI1) associated with atypical teratoid rabdoid tumor (AT / RT) from embryonal tumors, which is a subgroup of childhood central nervous system tumors.
Material and Method: The data of patients with AT / RT diagnosed between 2010 and 2015 in Ege University Medical Faculty were reviewed. Clinical findings and INI1 expression in tumor tissue using immunohistochemical analysis results of were evaluated. In all tumor tissues, all exonic regions of the hSNF5 / INI1 gene were amplified by PCR and after cheching by gel electrophoresis, DNA sequence analysis was performed. The results obtained were compared with the reference genome. The pathogenicity of the detected variants was assessed according to ACMG 2015 criteria. DNA sequencing in peripheral blood was performed in the patients with mutation in tumor tissue.
Results: Ten patients were enrolled in the study. 7 of them had AT/RT in central nervous system, 2 in soft tissue and 1 in renal. Of 7 patients who had AT / RT of the central nervous system (CNS), 4 (57%) were girls and 3 (43%) were boys. The median age at diagnosis was 18 months (3 months-12.9 years). 85% of our patients were diagnosed before 36 months. The mean survival time of the patients was 5.8 ( ± 28.7) months.
In 4 (57%) of the cases with AT/RT in CNS, the tumor was infratentorial, whereas in 3 (43%) the tumor was supratentorial. The median age at diagnosis was 19 months (16 months- 12,9 years) and the mean survival time was 7.3 (± 5.7) months in patients with tumors located in the supratentorial region. The median age at diagnosis was 17.5 months (3-19 months) and the mean survival time was 23.2 (± 38.5) months in the patients who were followed up for infratentorial tumor.
Gross total excision of 2 patients (28.5%) and partial excision of 5 patients (71.4%) were performed in 7 patients who were diagnosed with AT/RT of the CNS. The mean survival time of patients with gross total excision was 43.5 (± 53.7) months and the mean survival time of patients with residual tumor tissue was 5.8 (± 4.7) months.
The expression of hSNF5 / INI1 protein that has been examined by immunohistochemical methods in the patients with AT/RT of the CNS, soft tissue and renal rhabdoid tumors was evaluated and loss of protein expression was found in 77% of the patients.
All patients with soft tissue and renal rhabdoid tumors showed loss of hSNF5 / INI1 protein expression in tumor tissue.
Protein expression was lost in 4 of the 7 patients diagnosed with AT/RT of the CNS, protein expression was completely preserved in one patient, and focally conserved in one patient. In one patient, there was no data about protein expression.
The median age at diagnosis was 18 months (3-19 months) and the mean survival time was 3.2 (± 2.6) months in patients with loss of hSNF5 / INI1 protein expression.
In the hSNF / INI1 sequence analysis, mutations in tumor tissue were detected in 4 patients. Three of these patients had AT/RT of CNS and one of them had a soft-tissue rabboid tumor.
The median age at diagnosis was 19 months (17-19 months) and the mean survival time was 4 (± 2.6) months for patients with AT/RT of CNS with mutations in the hSNF5 / INI1 gene. The median age at diagnosis was 17 months (3 months-12,9 years) and the mean survival time was 25.7 (± 37.1) months in patients with no mutation in hSNF5 / INI1 gene.
There was a patient who was diagnosed with soft tissue rhabdoid tumor diagnosis and detected a mutation in the hSNF5 / INI1 gene in the tumor. At the time of diagnosis, the age of the patient is 3.5 months and the survival time is 15 months. The disease was remedied when this work was done. We have not detected mutation in the DNA obtained from the blood of patients who have mutation in the tumor.
Parents of mutation-bearing patients were evaluated using the Sanger sequence analysis for the same mutation. It was shown that the parents of the patients did not carry this mutation.
Conclusıons: Patients with AT / RT were found to have shorter survival times when mutations in the hSNF5 / INI1 gene were detected in the tumor. For this reason, mutation analysis could be recommended to predict prognosis. The future researchs conducted with larger patient groups is
needed to determine the role of molecular analysis in the patients with AT/RT. Unlike previous reports, no germline mutation in the hSNF5 / INI1 gene was detected in any of the patients with AT / RT. This result is thought to be related to the limited number of patients that could be analyzed.
Keywords: Central Nervous System tumors(CNS tumors),Atypical Teratoid/Rhabdoid Tumor (AT/RT), hSNF5/INI1
İÇİNDEKİLER
ÖNSÖZ VE TEŞEKKÜR ... ii
ÖZET ... iv
SUMMARY ... vi
TABLO LİSTESİ ... xii
ŞEKİL LİSTESİ ... xiii
KISALTMALAR ... xiv
1. GİRİŞ VE AMAÇ ... 1
2. GENEL BİLGİLER ... 4
2.1. Kanser Epidemiyolojisi ... 4
2.2. Çocukluk Çağında Santral Sinir Sistemi Tümörleri ... 6
2.2.1. Santral Sinir Sistemi (SSS) Tümörleri Epidemiyolojisi ... 6
2.2.2. Santral Sinir Sistemi (SSS) Tümörlerinde Risk Faktörleri ve Etyoloji ... 8
2.2.3. Santral Sinir Sistemi (SSS) Tümörlerinin Sınıflaması ... 11
2.2.4. Santral Sinir Sistemi (SSS) Tümörlerinde Klinik Bulgular ... 14
2.2.5. Santral Sinir Sistemi (SSS) Tümörlerinde Tanı Yöntemleri... 20
2.2.6. Santral Sinir Sistemi (SSS) Tümörlerinin Bir Alt Grubu Olan Atipik Teratoid Rabdoid Tümörün (AT/RT) Genel Özellikleri ... 21
2.3. Kanser ve Genetik ... 24
2.3.1. Kanser Genetiğinde Temel Kavramlar ... 24
2.3.2. Karsinogenez ... 29
2.4. AT/RT ve hSNF5/INI1 İlişkisi ... 30
2.4.1. hSNF/SWI’nın Hücre Bölünmesindeki Rolü ... 31
2.4.1. hSNF5/INI1 Tümör Supresör Geninin Keşfi ... 31
2.4.3. Bir Tümör Süpresör Gen Olarak Tanımlanan hSNF5/INI1 Mutasyonu ve AT/RT ... 34
3. GEREÇ VE YÖNTEM ... 35
3.1. Hasta Değerlendirmesi ... 35
3.2. hSNF/INI1 Geni Moleküler Analizi ... 35
3.3. Dizi Analizi Çalışması ... 36
3.3.1. PCR Reaksiyon Karışımı ... 36
3.3.2. Agaroz Jel Elektroforezi ... 36
3.3.3. PCR Ürünlerinin 1. Saflaştırılma İşlemi ... 37
3.3.4. Cycle Sequencing ... 38
3.3.5. İkinci Saflaştırma İşlemi (Zymo DNA Sequencing Clean-Up Kit) ... 40
3.3.6. Örneklerin Cihaza Yükleme Aşaması ... 40
3.3.7. Sonuçların Değerlendirilmesi ... 41 4. BULGULAR ... 42 5. TARTIŞMA ... 55 6. SONUÇLAR ... 72 KAYNAKLAR ... 75 EKLER ... 84 EK 1: Aile Ağaçları ... 84
Ek 2: Tümör Dokusunda Mutasyon Belirlenen Olguların Dizi Analizi Görüntüleri ... 86
Ek 3: Tümör Dokusundan Mutasyon Belirlenen Olguların Anne Baba Segragasyon Analizi Sonuçları ... 88
Ek 4: Olgu Rapor Formu ... 90
Ek-5: Genetik Çalışmalar İçin Bilgilendirilmiş Gönüllü Olur Formu ... 92
Ek 6: Bilgilendirilmiş Gönüllü Olur Formu ... 96
EK 7: Etik Kurul Onayı ... 101
Ek 8: Özgeçmiş Formu ... 104
TABLO LİSTESİ
Tablo 2.1. Çocukluk Çağında En Yaygın Görülen Maligniteler ... 4 Tablo 2.2. Türkiye ve ABD SEER Sonuçlarına göre Tümör Dağılımı... 6 Tablo 2.3. Çocuklarda SSS Tümörlerinin Yaş Gruplarına göre
Histolojik Dağılımı ... 7 Tablo 2.4. Pediatrik Beyin Tümörleri ile İlişkili Ailesel Sendromlar ... 10 Tablo 2.5. SSS Tümörlerinde İmmunohistokimyasal Özellikler ... 12 Tablo 2.6. Dünya Sağlık Örgütü Santral Sinir Sistemi Tümörleri
Sınıflaması ... 13 Tablo 2.7. SSS Tümörü Semptomlarının Yaş Gruplarına göre Özeti ... 15 Tablo 2.8. Çocuklarda SSS Tümörleri ile İlgili Belirti ve Bulguların
Görülme Sıklığı ... 16 Tablo 2.9. Çocuklarda SSS Tümörlerinin Yerleşim Yerlerine göre
Prezentasyonu ... 18 Tablo 2.10. Beyin Tümörü ile İlişkili Baş Ağrısı İçin “Kırmızı Bayrak”
Bulguları ... 19 Tablo3.1. Primerler ... 39 Tablo 4.1. SSS’nin AT/RT’ü, Yumuşak Doku ve Ekstrakranial
Yerleşimli Rabdoid Tümör Hastalarının Klinik,
İmmunohistokimyasal ve Moleküler Özellikleri ... 43 Tablo 4.2. hSNF5/INI1 Geninde Mutasyon Saptanan Hastaların
Mutasyon Özellikleri ... 47
ŞEKİL LİSTESİ
Şekil 2.1. 0-19 Yaşları Arasında SSS Tümörlerinin Histolojik
Tiplerine göre Dağılımı, CBTRUS 2007-2011. ... 8 Şekil 2.2. 0-19 Yaşları Arasında Primer Beyin ve SSS Tümörlerinin
Yerleşim Yerlerine göre Dağılımı, CBTRUS 2007-201120
Şekil 3.1. Agoroz jel görüntüsü ... 37 Şekil 4.1. Tümör Dokusunda INI1 Ekspresyon Kaybı Saptanan ve
PCR Başarısızlığı Gözlenen Hastaların Jel Elektroforezi
Görüntüsü ... 52 Şekil 4.2. INI1 Geninde Mutasyon Saptanan
HastalarınHistopatolojik ve İmmunohistokimyasal
Özellikleri ... 53
KISALTMALAR
ABD : Amerika Birleşik Devletleri
SEER : ABD Surveillance Epidemiology and End Results SSS : Santral Sinir Sistemi
ATRT : Atipik Teratoid Rabdoid Tümör PNET : Primitif Nöroektodermal Tümör NFP : Nöroflament Protein
GFAP : Glial Fibriler Asidik Protein SMA : Düz Kas Aktin
AFP : Alfa-fetoprotein
HCG : İnsan Koryonik Gonadotropin BOS : Beyin Omurilik Sıvısının BT : Bilgisayarlı Tomografi
MRG : Manyetik Rezonans Görüntüleme DSÖ : Dünya Sağlık Örgütü
GDP : Guanozin Difosfat GTP : Guanozin Trifosfat GTPaz : Guanozin Trifosfataz CDKs : Siklin Bağımlı Kinazlar TP53 : Tümör Protein p53 DNA : Deoksiribo Nükleik Asit RNA : Ribo Nükleik Asit
KML : Kronik Miyeloid Lösemi LOH : Loss Of Heterozigosite USG : Ultrasonografide
HIV : Human Immunodeficiency Virus / İnsan Bağışıklık Yetmezlik Virüsü
SWI : Switch
SNF : Sucrose Non-Fermenting IN : İntegraz Protein
1. GİRİŞ VE AMAÇ
Kanser kontrolsüz hücre çoğalmasıdır(1). Kanser yüzyıllardır varlığını sürdüren ve insan yaşamını tehdit eden bir hastalıktır. Malign tümörlerle ilgili tanımlara Mısır papirüsleri, Babil çivi yazısı tabletleri ve eski Hint yazmalarında rastlanılmaktadır. Kanser terimi ilk defa Hipokrat tarafından (M.Ö. 460-377) organizmanın şifa bulmayan yeni yapılanmaları için kullanılmıştır. Günümüzde kanser en basit anlatım ile kontrolsüz hücre çoğalması olarak tanımlanmaktadır. Varlığı insanlık tarihi kadar eskiye dayanan bu hastalığın epidemiyolojisi, etyolojisi, patogenezi, tanı ve tedavi yöntemleri ile ilgili her geçen gün yeni bilgiler keşfedilmektedir. Doğası gereği mortalitesi ve morbiditesi yüksek bir hastalık olması nedeni ile bilim adamları daha etkili tedaviler geliştirmek için son derece yoğun çalışmalar yürütmekte ve her geçen gün umut vadeden yeni tedavi yöntemleri geliştirilmektedir.
Dünyada son yıllarda “İnsan Genom Projesi” gibi genetik alanında büyük çalışmalardan elde edilen veriler ışığında hızlı gelişmeler kaydedildi. Bu sayede pek çok hastalığın genetik nedenleri ortaya koyuldu ve hedefe yönelik tedaviler keşfedildi. Kanser ile ilişkilipek çok tümör süpresör gen, protoonkogen ve onkogen tanımlandı. Kronik myeloid lösemi, meme kanseri gibi bazı kanser türlerinde moleküler genetik nedenlerin saptanması ve hedefe yönelik tedavi verilmesinin ardından son derece yüz güldürücü yanıtlar alındı. Tüm bu deneyimler nedeni ile “Kanser ve Genetik” ilişkisinin önemi her geçen gün artmaktadır.
En basit hali ile kontrolsüz hücre çoğalması olarak tanımlanan kanser çoğunlukla ileri yaşın hastalığı olarak bilinir. On dokuz yaş altında oldukça nadir görülmektedir. Amerika Birleşik Devletleri (ABD) verilerine göre yıllık insidansı 16,6 / 100000’dir. Çocukluk yaş grubunda görülen kanserler, tüm yaş grupları içinde bir yılda görülen tüm kanser vakalarının yaklaşık %1’ini oluşturmaktadır(2).
Çocukluk çağı kanserlerinin önemi yüksek mortaliteye sahip olması ile ilişkilidir. Malign neoplaziler 1-14 yaş arasındaki ölüm nedenlerinin başlarında yer almaktadır (2).
Gelişmiş ülkelerde çocukluk çağında en sık görülen üç malignite sırası ile lösemi, santral sinir sistemi (SSS) tümörleri ve lenfomadır. Ülkemizin de
içinde bulunduğu gelişmekte olan ülkelerde ise bu sıralama lösemi, lenfoma ve SSS tümörü şeklindedir(3).
Primer SSS tümörleri heterojen bir grup hastalık olup topluca ele alındığında çocukluk çağı kanserlerinin yaklaşık %20’sini oluşturmaktadır ve 0-14 yaşları arasında kanser ilişkili ölümlerin en sık nedenidir (3–5). İnsidansı 15 yaş altında 3,5/100000olarak bildirilmektedir, erkeklerde 1,2:1 oranında daha sık görülmektedir(6). SSS tümörlerinin mortalitesi 0-15 yaş arasındaki çocuklarda yaklaşık olarak %4,5 olarak belirtilmektedir (5).
Santral sinir sistemi (SSS) tümörlerinin etyolojisi iyi tanımlanamamakla birlikte bugüne kadar tüm kanserlerin etyolojisinde olduğu gibi santral sinir sistemi tümörlerinin etyolojisinde de genetik ve iyonize radyasyon gibi çevresel etkenler sorumlu tutulmaktadır (5).
Genetik nedenlerin çocukluk çağı SSS tümörleri etyolojisinde önemli bir yere sahipolduğunu düşündüren veriler mevcuttur. SSS tümörü olan olguların yaklaşık %5’ine nörofibromotoz, tuberoskleroz gibi herediter sendromlar eşlik etmektedir. Bazı familyal ve herediter sendromların beyin tümörlerinin artmış insidansıyla ilişkisi olduğu bilinmekle birlikte henüz patogenez ile ilişkili moleküler olaylar tam olarak bilinmemektedir (5).
Santral sinir sistemi (SSS) tümörlerinin sınıflaması daha çok hücre orijinine dayalı bir sınıflamadır. Zaman içerisinde meydana gelen gelişmeler sayesinde morfolojik değerlendirmenin yanında immunohistokimyasal ve moleküler genetik yöntemler de sınıflama da kullanılmaya başlamıştır.
Atipik teratoid rabdoid tümör (AT/RT) pediatrik SSS tümörlerinin nadir görülen bir alt tipidir. Sıklıkla üç yaşından küçük çocuklarda görülür ve oldukça malign bir tümördür. Tüm santral sinir sistemi tümörlerinin %1-2’sini oluştururken infant yaş grubundaki çocuklarda görülen SSS tümörlerinin %10’unu oluşturmaktadır(4).
Santral sinir sisteminin AT/RT’ü tanısı alan hastalarda 5 yıllık sağ kalım %39,5 olarak bulunmuştur (4). Bildirilen yaşam süreleri 0,5 ile 11 ay arasında değişmektedir (7,8).
Histolojik görünüm olarak AT/RT oldukça yoğun, son derece artmış selülerite gösteren andiferansiye hücrelerden oluşmaktadır. AT/RT’ün içerdiği rabdoid hücrelerin temel özellikleri, eksantrik yerleşimli çekirdeğe, belirgin çekirdekçiğe, eozinofilik sitoplazmaya ve eozinofilik inklüzyon cisimlerine
sahip olmalarıdır. Tümör dokusunda nekroz ve yüksek oranda mitotik aktivite sıklıkla görülmektedir (7). Bu histolojik özellikleri nedeni ile sıklıkla primitif nöroektodermal tümör (PNET), medulloblastom, germ hücreli tümör, koroid pleksus karsinomu ile karışabilmektedir(4,8).
SSS’nin AT/RT’ü son derece agresif, tedaviye yanıtı düşük, mortalitesi yüksek bir tümördür. Hastalar tanıdan kısa süre sonra kaybedildikleri için tedavide kullanılan cerrahi, kemoterapi, radyoterapiyöntemlerinin başarısını, uzun süreli etkilerini değerlendirmek için randomize kontrollü çalışmalar yapmak oldukça zordur. Bu yüzden bu konular ile ilgili bilgilerimiz kısıtlıdır.
Ayırıcı tanı, prognoz belirleme ve etkin tedavi yöntemleri geliştirme konusunda zorluklar yaşanan ve bu konularda çok sayıda kapsamlı çalışmaya ihtiyaç duyulan bu tümör ile ilgili hSNF5/INI1 genindeki genomik değişikliklerin bulunması ile yeni bir döneme girilmiştir.
Santral sinir sistemi (SSS), renal ve ekstrarenal yerleşimli rabdoid tümörlerin büyük çoğunluğunda hSNF5/INI1 geninde genomik değişiklikler olduğu gösterilmiştir. hSNF5/INI1 ile ilişkisi gösterilmiş olan ilk primer pediatrik beyin tümörü AT/RT’dür. hSNF5/INI1 geni zaman içinde AT/RT ile ilişkili aday bir tümör supresör gen olarak tanımlanmıştır (9). Anti-hSNF5/INI1 antikoru tanısal bir araç olarak kullanılmaya başlanmıştır ve yanlış tanı oranlarında düşüş sağlanmıştır. Ayırıcı tanı başarısında artış sağlaması ile birlikte pediatrik malign SSS tümörlerinde hSNF5/INI1 protein ekspresyonu rutin olarak analiz edilmeye başlamıştır (4).
Bu çalışmada, çocukluk çağı santral sinir sistemi tümörlerinin bir alt grubu olan embriyonel tümörlerden, atipik teratoid rabdoid tümör (AT/RT), tanısı almış olan hastalarda, bu tümör ile ilişkili olan bir tümör supresör gen (hSNF5/INI1) gen mutasyonunun araştırılması amaçlanmıştır.
2. GENEL BİLGİLER
2.1.Kanser Epidemiyolojisi
En basit tanımı ile kontrolsüz hücre çoğalması olarak tanımlanan kanser çoğunlukla ileri yaşın hastalığı olarak bilinir. 19 yaş altında oldukça nadir görülmektedir.Amerika Birleşik Devletleri (ABD) verilerine göre yıllık insidansı 16,6/100000’dir. Çocukluk yaş grubunda görülen kanserler, tüm yaş grupları içinde bir yılda görülen tüm kanser vakalarının yaklaşık %1’ini oluşturmaktadır (2). Tüm yaş gruplarında 5 yıllık yaşam oranları 1977’de %61 iken 2005’te %81,6’ya yükselmekle beraber malign neoplaziler halen 1-14 yaş arasındaki ölüm nedenlerinin başlarında yer almaktadır. ABD’nde 15 yaş altındaki çocuklarda bir yılda 1500-1600 çocuk kanser nedeni ile hayatını kaybetmektedir(2).
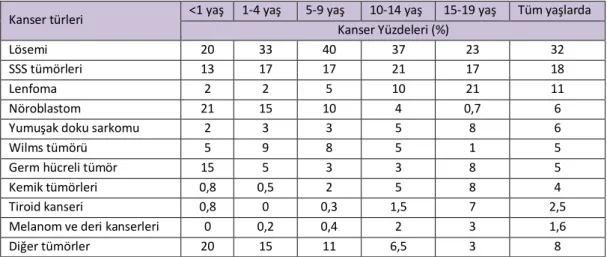
ABD verilerine göre çocukluk çağı kanserlerinin %40’ını lenfohemotopoetik kanserler, %30’unu sinir sistemi kanserleri, %10’unu embriyonel tümörler ve sarkomlar oluşturmaktadır. Çocukluk yaş grubunda, erişkinlerde sık rastladığımız akciğer, meme, kolon, prostat gibi organların epitelyal tümörlerine nadiren rastlanmaktadır(2).
Tablo 2.1. Çocukluk Çağında En Yaygın Görülen Maligniteler
Kanser türleri <1 yaş 1-4 yaş 5-9 yaş 10-14 yaş 15-19 yaş Tüm yaşlarda Kanser Yüzdeleri (%)
Lösemi 20 33 40 37 23 32
SSS tümörleri 13 17 17 21 17 18
Lenfoma 2 2 5 10 21 11
Nöroblastom 21 15 10 4 0,7 6
Yumuşak doku sarkomu 2 3 3 5 8 6
Wilms tümörü 5 9 8 5 1 5
Germ hücreli tümör 15 5 3 3 8 5
Kemik tümörleri 0,8 0,5 2 5 8 4
Tiroid kanseri 0,8 0 0,3 1,5 7 2,5
Melanom ve deri kanserleri 0 0,2 0,4 2 3 1,6
Diğer tümörler 20 15 11 6,5 3 8
The most common causes of cancer based upon age in the United States (2000 to 2011) from data obtained by the Surveillance, Epidemiology, and End Results program, National Cancer Institute.
Reproduced from: Surveillance, Epidemiology, and End Results (SEER) Program (www.seer.cancer.gov) SEER*Stat Database: Incidence - SEER 18 Regs Research Data + Hurricane Katrina Impacted Louisiana Cases, Nov 2013 Sub (2000-2011) <Katrina/Rita Population Adjustment> - Linked To County Attributes - Total U.S., 1969-2012 Counties, National Cancer Institute, DCCPS, Surveillance Research Program, Surveillance Systems Branch, released April 2014 (updated 5/7/2014), based on the November 2013 submission.
Çocukluk yaş grubunda kanser hastalığının erken çocukluk çağında ve adölesan dönemde olmak üzere iki pik yaptığı görülmektedir. Yaşamın birinci yılı içinde en sık nöroblastom, nefroblastom, retinoblastom, rabdomyosarkom, hepatoblastom ve medulloblastom gibi embriyonel tümörler görülürken 2-5 yaş arasında akut lösemi, hodgkin dışı lenfoma ve gliom insidansı pik yapmaktadır. Kemik maligniteleri, hodgkin lenfoma, germ hücreli tümörlerin görülme sıklığı 10 yaşından sonra artış göstermektedir(2).Çocukluk çağında en yaygın görülen malignitelerin yaş gruplarına göre dağılımı Tablo 2.1’de verilmiştir.
Türkiye'de çocukluk çağı kanserlerinin epidemiyoloji, insidansı ve yaşam hızlarına yönelik veriler oldukça sınırlıdır. Türk Pediatrik Onkoloji Grubunun 33 merkezden topladığı 2002 yılı verilerine göre 1073 vakanın kaydı yapılmıştır. Bu kayıtlara göre tüm vakalar için ortanca yaş 6,4 olup, erkek kız oranı 1,39 olarak bulunmuştur. 33 merkezden bildirilen 1073 vakanın 1 yıllık yaşam hızı %77 olarak bulunmuştur(3). Gelişmiş ülkelerde uzun süreli yaşam hızlarının %75-80’lere ulaştığı bilinmektedir. Gelişmiş ülkelerin gerisinde kalan ülkemizde bu verilerin iyileştirilmesi gerekmektedir. Bu kayıtlardan elde edilen önemli sonuçlardan bir diğeri ülkemizde gelişmiş ülkelerle karşılaştırıldığında lenfomaların santral sinir sistemi tümörlerinden daha sık görülmesidir(3).Türkiye'de ve Amerika'da yaşayan çocuklardaki kanser dağılımı Tablo 2.2’de karşılaştırmalı olarak verilmiştir.
Gelişmiş ülkelerde santral sinir sistemi (SSS) tümörleri lösemilerden sonra ikinci sırada yer alırken ülkemizde ve diğer gelişmekte olan ülkelerde lösemi ve lenfomalardan sonra üçüncü sırada yer almaktadır. SSS tümörleri çocukluk çağında görülen en sık solid tümör tipidir.
Tablo 2.2. Türkiye ve ABD SEER Sonuçlarına göre Tümör Dağılımı
Tümör tipi Türkiye (%) SEER(%)
Lösemiler 31,9 25,8
Lenfomalar 16,8 13,3
Beyin ve spinal kanal tümörleri 13,4 23,5
Sempatiksistem tümörleri 7,4 4,1
Retinoblastom 3,2 1,7
Böbrek tümörleri 5,4 3,4
Karaciğer tümörleri 1,4 1,3
Kemik tümörleri 6,0 4,6
Yumuşak doku sarkomları 6,4 6,4
Gonad ve germ hücreli tümörler 4,6 6,3
Epitelyal tümörler 2,8 8,9
Diğer malign neoplaziler 0,6 0,2
SEER: ABD Surveillance Epidemiology and End Results
2.2. Çocukluk Çağında Santral Sinir Sistemi Tümörleri
2.2.1. Santral Sinir Sistemi (SSS) Tümörleri Epidemiyolojisi
Primer SSS tümörleri heterojen bir grup hastalık olup topluca ele alındığında çocukluk çağı kanserlerinin yaklaşık %20’sini oluşturmaktadırve SSS tümörleri 0-14 yaşları arasında kanser ilişkili ölümlerin en sık nedenidir(3–5). Gelişmiş ülkelerde çocukluk çağı kanserleri arasında ikinci sıklıkta görülmektedir. Ülkemizde ve diğer gelişmekte olan ülkelerde, gelişmiş ülkelerden farklı olarak görülme sıklığı açısından üçüncü sırada yer almaktadır(3,5). İnsidansı 15 yaş altında 3,5/100000olarak bildirilmektedir. Farklı bölgelerde oldukça benzer bir insidansa sahiptir. 0-15 yaş arasındaki çocuklarda İsveç'teki 35,9/1000000, Japonya'da 36.1/1000000, İngiltere'de 32.7/1000000, ABD’nde 47,1/1000000 olduğu değişik bölgelerdeki çeşitli çalışmalarda gösterilmiştir(6).
0-15 yaş arasında SSS tümörleri mortalitesi %4,5’e yaklaşmaktadır(5). SSS tümörlerinin insidansı hayatın ilk on yılında bir zirve yaparken ikinci dekatta azalma göstermektedir ve yedinci dekatta ikinci zirvesini yapmaktadır. Malign SSS tümörleri insidansı %55’e karşılık %45 oranla erkeklerde kızlardan daha yüksektir. Bu farkın büyük kısmı, primitif nöroektodermal tümör (PNET), medulloblastom ve ependimomaların
erkeklerde daha sık görülmesinden kaynaklanmaktadır. Medulloblastom ve ependimom dışındaki diğer santral sinir tümörlerinde erkek ve kız oranları yaklaşık olarak eşittir(10).
Tablo 2.3.Çocuklarda SSS Tümörlerinin Yaş Gruplarına göre Histolojik Dağılımı
Histolojik Tip 0-14 yaş (%) 15-19 yaş (%)
Pilositik astrositom 20,5 14,6 Diğer tüm astrositomlar 9,5 10,4 Embriyonel tümörler 15,6 5,6 Glioblastom 2,8 3,8 Epandimom 6,4 5 Oligodendrogliom 1,3 2,4 Pitüiter 1,5 13,9 Kraniyofaringioma 3,1 2,4
Germ hücreli tümörler 3,6 4,9
Diğer 35,7 36,9
Tümörlerin primer yerleşim yerleri yaş ile ilişkili farklılıklar göstermektedir. İki yaşından adolesan yaş grubuna kadar olan çocuklarda üçte iki oranında posterior fossa tümörleri görülürken iki yaşından küçük çocuklarda ve adolesan dönemde supratentorial tümörler ile posterior fossa tümörleri eşit oranlarda görülmektedir. On yaş altındaki çocuklarda SSS tümörleri en sık serebellumda yerleşmektedir (10). Pediatrik SSS tümörlerinin histolojik alt tiplerinin yaşa göre dağılımı Tablo 2.3’de verilmiştir.
0-14 yaş arasındaki çocuklarda en sık pilositik astrositom ve PNET, medulloblastom görülürken adolesanlarda en sık pitüiter tümörler ve pilositik astrositom görülmektedir (5). Çocukluk çağı SSS tümörlerinin histolojik tiplere göre dağılımı Şekil 2.1’dedir. Erişkin yaş grubunda görülen yüksek ‘grade’li gliomlar ve gliablastome multiforme ise bu yaş grubunda oldukça nadir görülmektedir. Bunların yerini embriyonal histolojili tümörler almaktadır(5).
Şekil 2.1. 0-19 Yaşları Arasında SSS Tümörlerinin Histolojik Tiplerine göre Dağılımı,
CBTRUS 2007-2011.
2.2.2. Santral Sinir Sistemi (SSS) Tümörlerinde Risk Faktörleri ve Etyoloji
Daha önce yapılan etiyolojik araştırmalarda pediatrik SSS tümörlerinin diğer çocukluk çağı kanserlerinden ayrı bir antite olduğu düşünülerek tasarlanan bir çalışma olmadığı için SSS tümörlerinin etyolojisine yönelik bilgilerimiz diğer çocukluk çağı kanserleri ile ilgili tecrübelerimize dayanmaktadır. Bu nedenle etyolojisi iyi tanımlanamamakla birlikte bugüne kadar tüm kanserlerin etyolojisinde olduğu gibi santral sinir sistemi
Oligodendrogliom 1% Meningiom 3% Glioblastom 3% Kraniofaringiom 4% Germ hücreli tümör 4% Sinir kılıf tümörleri 5% Epandimal tümörler 5% Nöronal ve mixt nöroglial tümörler 7% Diğer astrositomlar 9% Pitiuter bölge tümörleri 10% Diğer 10% Malign gliom 12% Embriyonal tümörler 12% Pilositik astrositom 15% Oligoastrositik tümörler 0% Lenfoma 1%
Reproduced with permission from: CBTRUS Statistical Report: NPCR and SEER, 2007-2011. Copyright ©2014 Central Brain Tumor Registry of the United States.
tümörlerinin etyolojisinde de genetik, iyonize radyasyon gibi çevresel etkenler sorumlu tutulmaktadır(5).
Kranial iyonize radyasyona maruziyet SSS tümörlerinin yüksek insidansı ile ilişkilidir(5). 1940’lı yıllarda tinea capitis nedeniyle iyonize radyasyon tedavisi almış çocuklarda gliom, meningiom ve sinir kılıfı tümörleri için ortalama 22–34 yıl sonra artmış risk saptanmıştır. Son zamanlarda akut lenfositik lösemi nedeni ile proflaktik kraniyal radyoterapi alan hastalarda ileri dönemlerde SSS tümörlerinin ortaya çıktığı tanımlanmıştır (11,12).
Çok sayıda çalışmada SSS tümörleri ile çevresel etkenlerin birliktelik açısından ilişkisiz olduğu sonucuna varılmış olmasına rağmen aksini gösteren çalışmalar da mevcuttur. Solventlere ve polisiklik aromatik hidrokarbonlara maruziyetin özellikle PNET ve glial tümörler olmak üzere beyin tümörü gelişme riskini önemli ölçüde artırdığı gösterilmiştir. Benzer şekilde kimya endüstrisinde çalışanların çocuklarında SSS tümör riskinin arttığı gösterilmiştir (13).
Evde kullanılan kimyasal pestisitlerin içeriğinde bulunan N-nitroso bileşiklerinin hayvan modellerinde nörokarsinojen olduğu gösterilmiştir. Bileşiğin bu etkiyi Ras onkogen aktivasyonuna yol açarak yaptığı düşünülmektedir. Çocukluk çağı astrositomlarında da Ras onkogen mutasyonunun gösterilmesi çevresel maruziyet ile SSS tümörü gelişimi arasında bir ilişki olabileceğini destekler niteliktedir(13).
Vitamin C, E ve minerallerin antioksidan etkileri sayesinde DNA yapısını serbest radikallere karşı koruyarak kansere karşı önleyici etki gösterdikleri düşünülmektedir (13).
Genetik nedenler çocukluk çağı SSS tümörleri etyolojisinde önemli bir yere sahiptir ancak henüz patogenez ile ilişkili moleküler yolaklar tam olarak bilinmemektedir. Bazı familyal ve herediter sendromların beyin tümörlerinin artmış insidansıyla ilişkisi olduğu bilinmektedir ve SSS tümörü olan olguların yaklaşık %5’inde herediter sendrom mevcuttur. Nörofibromatozis tip 1, Nörofibromatoz tip 2, von Hippel Lindau, Tüberoskleroz, Li-Fraumeni, Cowden, Turcot, Nevoid bazal hücreli karsinom sendromları SSS tümörleri ile ilişkileri iyi bilinen sendromlardır (5). Ataksi telenjiektazi, Gorlin sendromu, familyal adenomatozis polipozis, herediter nonpolipozis kolorektal kanser, multiple endokrin neoplazi tip 1 sendromları da SSS tümörleri ile ilişkili
oldukları gösterilmiş olan sendromlardır (14). Pediatrik beyin tümörleri ile ilişkili ailesel sendromlar ve bu sendromlar ile ilişkili genler Tablo 2.4’tedir.
Tablo 2.4. Pediatrik Beyin Tümörleri ile İlişkili Ailesel Sendromlar
Sendrom Merkezi Sinir Sistemi Bulguları Kromozom Gen
Nörofibromatozis tip 1 (OD)
Optik yol gliomları, astrositom, malign periferik sinir kılıf tümörleri,
nörofibromlar
17q11 NF1
Nörofibromatozis tip 2 (OD)
Vestibüler schwannomlar, meningiomlar, spinal kord epandimomu ve astrositomu, hamartomlar 22q12 NF2 Von Hippel-Lindau (OD) Hemanjioblastom 3p25-26 VHL Tuberosklerozis (OD)
Subepandimal dev hücreli astrositom, kortikal tuberler
9q34 TSC1
16q13 TSC2 Li-Fraumeni
(OD)
Astrositom, primitif nöroektodermal
tümörler 17q13 TP53
Cowden (OD)
Serebellumun displastik gangliositomu
(Lhermitte- Duclos hastalığı) 10q23 PTEN Turcot
(OD)
Medulloblastom 5q21 APC
Glioblastom 3p21 hMLH1
7q22 hPSM2 Nevoid Bazal Hücreli
Karsinom (OD) Medulloblastom 9q31 PTCH
Bu sendromların çoğu kalıtsal hastalıklardır ve bu hastalıklar ile ilişkili genetik bozuklukları tanımlamak bize karsinogeneze yol açan moleküler yolakları anlama imkanı sunmaktadır. Böylece gelecekte bu moleküler yolakları hedef alan özgün tedaviler keşfetmenin yolu açılmaktadır.
Beyin tümörlerinin birçoğunun altında yatan somatik mutasyonların nedenleri bilinmese de çocukluk çağında SSS tümörleri ile tipik ilişkisi olduğu düşünülen genetik anormallikler giderek artan kanıtlarla gösterilmektedir. Somatik veya germline mutasyonlar, kromozomal kırıklar, kromozomal aberasyonlar, heterozigozite kaybı gibi mekanizmalar ile bazı kanserlere yatkınlık oluştuğu artık kabul edilen bir gerçektir. En belirgin genetik anormallikler ependimom, medulloblastom/PNET ve atipik teratoid rabdoid tümörlerde (AT/RT) gösterilmiştir. Medulloblastom, ependimom ve AT/RT’demonosomi 22; ependimomlarda da 6q delesyonu ile birliktelik saptanmıştır. 22q11.2’de bulunan hSNF5/INI1 geni santral sinir sistemi ve santral sinir sistemi dışı AT/RT’lerde tespit edilmiştir(9,15–21).
2.2.3. Santral Sinir Sistemi (SSS) Tümörlerinin Sınıflaması
SSS tümörlerinde bugüne kadar çok sayıda sınıflama kullanılmıştır. SSS tümörlerinde kullanılan ilk sınıflamalar daha çok hücre orjinine dayalı sınıflamalardır. Zaman içerisinde kanserin genetik bir hastalık olduğunun anlaşılması ile birlikte sınıflama yapılırken morfolojik değerlendirme kadar histogenetik kavramların da göz önünde bulundurulması gerektiği anlaşılmıştır.
Hücre tiplerini belirleme de immunohistokimyasal ve moleküler biyolojik tekniklerin kullanıldığı fenotipik yaklaşım objektif bir tanımlama getirmesi açısından kullanılan bir diğer yöntemdir. Bu yöntemde hücre iskeleti, membran proteinleri, hormonal polipeptitler ve nörotransmitterler gibi spesifik antijenlere yönelik monoklonal antikorlar kullanılmaktadır. Bu tarz bir fenotipik yaklaşım morfolojik olarak büyük benzerlik gösteren atipik teratoid rabdoid tümörün (AT/RT), PNET, medulloblastomdan ayrımında oldukça yardımcı olmaktadır (7).
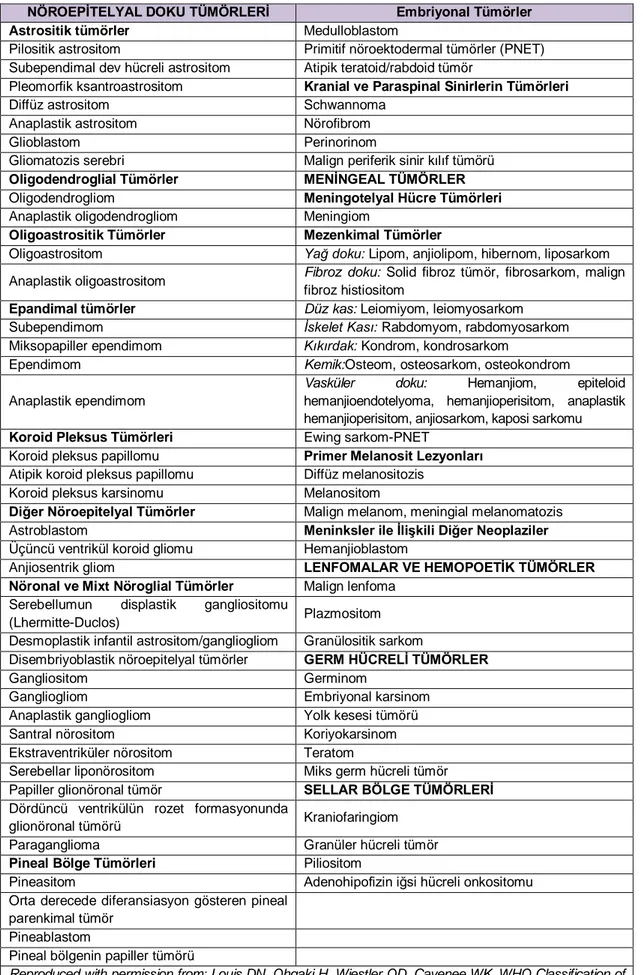
Günümüzde Dünya Sağlık Örgütünün (DSÖ) yaptığı sınıflama kullanılmaktadır. DSÖ’nün SSS tümörleri ile ilgili yaptığı en yeni sınıflama 2016’da yayınlanmıştır, sınıflama Tablo 2.6’te verilmiştir. Yapılan son sınıflamada moleküler parametrelerin sınıflamaya eklenmesi ile mikroskobik incelemeye dayanan asırlık tanı prensiplerinde büyük değişiklikler olmuştur (22).
2016’da yayınlanan son sınıflamaya göre artık immunohistokimyasal yöntemler dışında moleküler genetik yöntemler de sınıflamayı etkilemektedir. Bunun en çarpıcı örneğini AT/RT sınıflamasında karşımıza çıkmaktadır. AT/RT hastalarında hSNF5/INI1 geninde alterasyonlar/değişiklikler olduğu bilinmektedir. Bu değişiklikler, INI1’e karşılık gelen proteinlerin tümör dokusunda immünohistokimyasal yöntemler kullanılarak tespit edilmesiyle gösterilebilir. Bu genetik değişikliğin gösterilmesi artık AT/RT için tanısal önem taşımaktadır. Bir tümörün AT/RT'nin histolojik özelliklerine sahip olması ancak hSNF5/INI1 genindeki değişikliği taşımaması halinde bu tümör sadece rabdoid özellikleri olan SSS embriyonal tümörü olarak tanımlanabilmektedir. Bir diğer ifade ile AT/RT tanısı için artık bu karakteristik moleküler defektin objektif olarak gösterilmesi gerekmektedir (22).
Tablo 2.5. SSS Tümörlerinde İmmunohistokimyasal Özellikler Glial fibriler asidik
protein (GFAP)
Astrositom, ependimom, mikst gliom, gliosarkom, gangliogliom, glioblastoma multiforme, gliofibrom; nadiren oligodendrogliom, kapiller hemanjioblastom ve koroid pleksus papillomu, PNET ve AT/RT
Nörofilament protein (NFP)
Gangliogliom, gangliositom, PNET, nörositom, subependimal dev hücreli astrositom ve AT/RT
Vimentin
Mezenşimal tümörler, meninjiomlar, sarkomlar, melanomlar, lenfoma, ependimom, astrositom, gliofibrom, kordoma, schwannom, hemanjioblastom, karsinom, PNET ve AT/RT
Desmin Kas dokusu içeren tümörler ( rabdomyosarkom, teratom, vs.) ve
PNET
Sitokeratin
Kordoma, koroid pleksus tümörleri, meningiom, bazı anaplastik gliomlar, nongerminamatöz germ hücreli tümörler, PNET ve AT/RT
Epitelyal membran
antijeni Meningiom, ependimom, epitelyal teratom alanları ve AT/RT Sinaptofizin PNET, gangliogliom, gangliositom, santral nörositom ve
nöroendokrin tümörler
Düz kas aktin (SMA) Kas içeren tümörler ve AT/RT
Alfa-fetoprotein
(AFP) Embriyonal karsinom ve endodermal sinüs tümörü İnsan koryonik
gonadotropin (HCG) Germinom, koryokarsinom
Rabdoid tümörler benzer şekilde oval, orta boy, tipik eksantrik nükleuslu hücrelere sahiptir. PNET, medulloblastom ve nadiren de koroid pleksus karsinomu gibi tümörlerden immunhistokimyasal yöntemler kullanılarak vimentin, epitelyal membran antijeni, SMA, NFP, GFAP ve keratin eksprese etmeleri ile ayrılmaktadırlar. İmmunoperoksidaz tekniklerinin kullanılması ile de astrositomlar; subependimal dev hücreli astrositom, pleomorfik ksantoastrositom ve süperfisiyal serebral astrositom gibi daha spesifik alt gruplara ayrılmaktadır.
SSS tümörlerinde tanıda immunohistokimyasal olarak sık kullanılan tümör belirleyicilerini Tablo 2.5’te özetlenmiştir.
Tablo 2.6.Dünya Sağlık Örgütü Santral Sinir Sistemi Tümörleri Sınıflaması
NÖROEPİTELYAL DOKU TÜMÖRLERİ Embriyonal Tümörler Astrositik tümörler Medulloblastom
Pilositik astrositom Primitif nöroektodermal tümörler (PNET)
Subependimal dev hücreli astrositom Atipik teratoid/rabdoid tümör
Pleomorfik ksantroastrositom Kranial ve Paraspinal Sinirlerin Tümörleri
Diffüz astrositom Schwannoma
Anaplastik astrositom Nörofibrom
Glioblastom Perinorinom
Gliomatozis serebri Malign periferik sinir kılıf tümörü
Oligodendroglial Tümörler MENİNGEAL TÜMÖRLER
Oligodendrogliom Meningotelyal Hücre Tümörleri
Anaplastik oligodendrogliom Meningiom
Oligoastrositik Tümörler Mezenkimal Tümörler
Oligoastrositom Yağ doku: Lipom, anjiolipom, hibernom, liposarkom
Anaplastik oligoastrositom Fibroz doku: Solid fibroz tümör, fibrosarkom, malign fibroz histiositom
Epandimal tümörler Düz kas: Leiomiyom, leiomyosarkom
Subependimom İskelet Kası: Rabdomyom, rabdomyosarkom
Miksopapiller ependimom Kıkırdak: Kondrom, kondrosarkom
Ependimom Kemik:Osteom, osteosarkom, osteokondrom
Anaplastik ependimom
Vasküler doku: Hemanjiom, epiteloid
hemanjioendotelyoma, hemanjioperisitom, anaplastik hemanjioperisitom, anjiosarkom, kaposi sarkomu
Koroid Pleksus Tümörleri Ewing sarkom-PNET
Koroid pleksus papillomu Primer Melanosit Lezyonları
Atipik koroid pleksus papillomu Diffüz melanositozis
Koroid pleksus karsinomu Melanositom
Diğer Nöroepitelyal Tümörler Malign melanom, meningial melanomatozis
Astroblastom Meninksler ile İlişkili Diğer Neoplaziler
Üçüncü ventrikül koroid gliomu Hemanjioblastom
Anjiosentrik gliom LENFOMALAR VE HEMOPOETİK TÜMÖRLER
Nöronal ve Mixt Nöroglial Tümörler Malign lenfoma Serebellumun displastik gangliositomu
(Lhermitte-Duclos) Plazmositom
Desmoplastik infantil astrositom/gangliogliom Granülositik sarkom
Disembriyoblastik nöroepitelyal tümörler GERM HÜCRELİ TÜMÖRLER
Gangliositom Germinom
Gangliogliom Embriyonal karsinom
Anaplastik gangliogliom Yolk kesesi tümörü
Santral nörositom Koriyokarsinom
Ekstraventriküler nörositom Teratom
Serebellar liponörositom Miks germ hücreli tümör
Papiller glionöronal tümör SELLAR BÖLGE TÜMÖRLERİ
Dördüncü ventrikülün rozet formasyonunda
glionöronal tümörü Kraniofaringiom
Paraganglioma Granüler hücreli tümör
Pineal Bölge Tümörleri Piliositom
Pineasitom Adenohipofizin iğsi hücreli onkositomu
Orta derecede diferansiasyon gösteren pineal parenkimal tümör
Pineablastom
Pineal bölgenin papiller tümörü
Reproduced with permission from: Louis DN, Ohgaki H, Wiestler OD, Cavenee WK. WHO Classification of Tumours of the Nervous System. IARC Press, Lyon 2007. Copyright © 2007 IARC Press.
2.2.4. Santral Sinir Sistemi (SSS) Tümörlerinde Klinik Bulgular
Çocukluk çağı SSS tümörlerinespesifik olan tanıda yol gösterici olabilecek hiçbir patognomonik klinik bulgu yoktur. Bulantı, kusma, huzursuzluk gibi semptomların öncelikle çocukluk çağında çok daha sık görülen gastrointestinal bozukluklar gibi basit nedenlerden kaynaklandığının düşünülmesi ve bu şekilde yönetilmesi tanıda gecikmelere neden olabilir (23). Tüm kanserlerde olduğu gibi SSS tümörlerinde de erken tanı prognoz açısından belirleyici öneme sahip olduğu için bu hasta grubunda hastaneye başvuru yakınmalarını iyi bilmemiz gerekmektedir.
Kliniğe başvuruyakınması çoğunlukla tümörün yerleşim yerine çocuğun yaşına, gelişim düzeyine ve tümör tipine göre değişkenlik göstermektedir. Yaş gruplarına SSS tümörü semptomları Tablo2.7’de özetlenmiştir. SSS tümörlerine ait ilk bulgu olarak genellikle beyin omurilik sıvısının (BOS) drenaj yollarında obstürksiyona bağlı gelişen kafa içi basınç artışı bulgularını görülmektedir. Hastalığın seyri sırasında nörolojik ve sistemik fonksiyon bozuklukları gelişmektedir (5,24,25).
Başlangıç bulguları çoğunlukla nonspesifik ve sinsi seyirli olduğu için tümör belirli boyutlara ulaşmadan tanı konulması oldukça güçtür. Semptomun başlaması ile teşhis arasındaki süre düşük evreli tümörlerde uzun sürerken, yüksek evreli tümörlerde daha kısa sürmektedir (5).
Tümör yerleşim yerine bağlı fokal nörolojik bulgu, epilepsi, düşünce ve davranış bozukluğu görülebilmektedir. En sık klasik triad olan baş ağrısı, bulantı ve kusma görülmektedir (5,24,25).
Tablo 2.7. SSS Tümörü Semptomlarının Yaş Gruplarına göre Özeti
Yaş grupları Semptomlar
< 5 yaş
• Persistan/tekrarlayan kusma
• Denge, koordinasyon, yürüme problemi • Anormal göz hareketleri
• Davranış değişikliği, letarji • Nöbet (ateşsiz dönemde)
• Anormal baş pozisyonu (tortikollis, ense sertliği)
5-11 yaş
• Persistan/tekrarlayan baş ağrısı • Persistan/tekrarlayan kusma
• Denge, koordinasyon, yürüme problemi • Anormal göz hareketleri
• Bulanık veya çift görme • Davranış değişikliği • Nöbet
• Anormal baş pozisyonu (tortikollis, ense sertliği)
12-18 yaş
• Persistan/tekrarlayan baş ağrısı • Persistan/tekrarlayan kusma
• Denge, koordinasyon, yürüme problemi • Anormal göz hareketleri
• Bulanık veya çift görme • Davranış değişikliği • Nöbet
• Anormal baş pozisyonu (tortikollis, ense sertliği) • Pubertede gecikme veya duraklama, büyüme geriliği (Royal College of Paediatrics and Child Health et al 2011)
Tablo 2.8. Çocuklarda SSS Tümörleri ile İlgili Belirti ve Bulguların Görülme Sıklığı İnfant ve 4 yaşın altındaki çocuklar Tüm çocuklar (daha büyük çocuklar ve adölesanlar dahil)
Makrosefali (%41) Baş ağrısı (%33)
Bulantı ve kusma (%30) Bulantı ve kusma (%32)
İrritabilite (%24) Anormal yürüyüş ve denge (%27)
Letarji (%21) Nöbet (%13)
Anormal yürüyüş ve denge (%19) Papilödem (%13)
Kilo kaybı/ büyüme geriliği (%14) Kafa içi basınç artışına ait bulgular (%10) Fontonelde bombelik ve/veya sütürlerde
açılma (%13) Şaşılık (%7)
Nöbet (%10) Davranış değişikliği veya okul başarısında gerileme (%7)
Papilödem (%10) Makrosefali (%7)
Baş ağrısı (%10) Kranial sinir paralizisi (%7) Fokal nörolojik bulgular (%10) Letarji / yorgunluk (%6) Kafa içi basınç artışına ait bulgular (%9) Anormal göz hareketleri (%6) Fokal motor güçsüzlük (%7) Hemipleji (%6)
Baş eğme (%7) Bilinç düzeyinde değişiklik (%5) Bilinç düzeyinde değişiklik (%7) Kilo kaybı (%5)
Şaşılık (%6) Görme bozuklukları (%5)
Anormal göz hareketleri (%6)
Gelişim geriliği veya kazanılmış becerilerin kaybı (%5)
Hemipleji (%5)
1. Wilne SH, Dineen RA, Dommett RM, et al. Identifying brain tumours in children and
young adults. BMJ 2013; 347:f5844.
2. Wilne S, Collier J, Kennedy C, et al. Presentation of childhood CNS tumours: a
systematic review and meta-analysis. Lancet Oncol 2007; 8:685.
Kafa içi basınç artışına ait semptomlar okul çocuklarında okul başarısında azalma, yorgunluk, aralıklı baş ağrıları, giderek artış gösteren sabahları daha yoğun olan baş ağrıları, konuşmada, kişilikte değişiklikler, kusma, letarji şeklinde kendini gösterirken küçük çocuklarda ve infantlarda irritabilite, iştahsızlık, büyüme ve gelişme geriliği, makrosefali, gözlerde batan güneş manzarası, kranial sinir felçlerine bağlı göz hareketlerinde kısıtlılık, nörojenik ağlama, kusma olarak karşımıza gelebilmektedir (5,23,25).
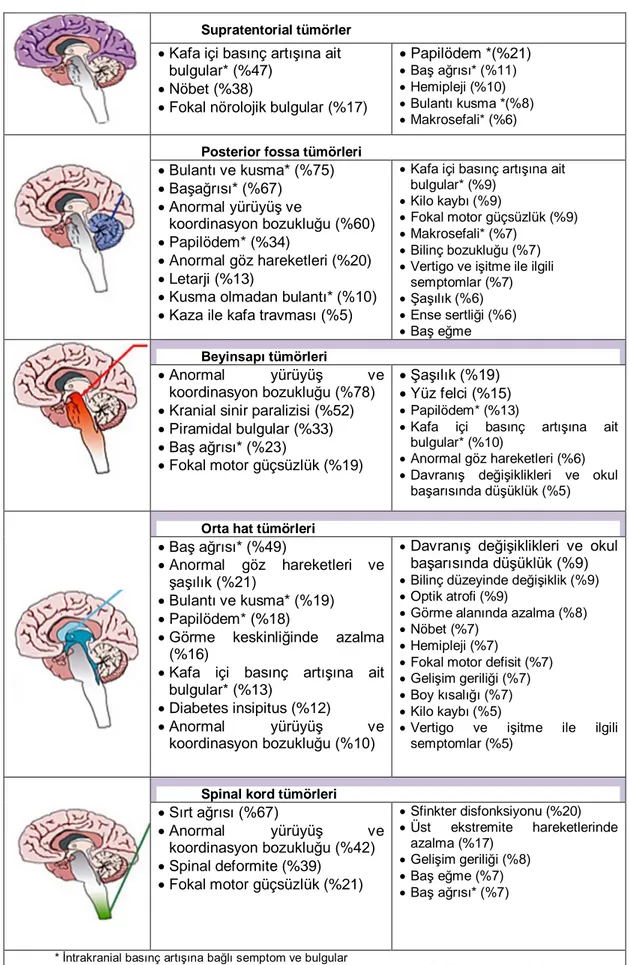
Semptomlar bize tümörün yerleşimi ile ilgili ipucu verebilmektedir. Motor güçsüzlük, duysal değişiklikler, kişilik, zekadaki değişiklikler, konuşmada bozulma, nöbet ve refleks anormallikleri daha çok supratentoriyal (kortikal) lezyonlarda görülmektedir. Klasik triad olan bulantı, kusma, baş ağrısının yanında papil ödem infratentoriyal ve orta hat tümörleriyle ilişkilidir. Bulanık görme, diplopi, nistagmus, denge, yürüyüş ve koordinasyon bozuklukları
infratentoriyal tümörlerde görülmektedir. Bakış paralizisi, multiple kraniyal sinir paralizileri, motor nöron defisitleri beyin sapı lezyonlarında görülen bulgulardır. Suprasellar bölge tümörleri diyabet insipitus, galaktore, puberte prekoks, hipotiroidi gibi nöroendokrin bozukluklarla kendini gösterebilmektedir. Spinal kord tümörleri ve beyin tümörlerinin spinal korda yayılımı motor ve duysal defisitler, barsak ve mesane defisitleri, sırt ağrısı şeklinde bulgu verebilmektedir (5). Çocuklarda SSS tümörlerinin yerleşim yerlerine göre prezentasyonu Tablo 2.9’da özetlenmiştir.
Hipotalamik bölge tümörlerinde diensefalik sendrom olarak tanımlanmış olan iştah artışı olmasına rağmen görülen aşırı kilo kaybı, gelişme geriliği, öforik duygu durumu görülmektedir. Diensefalik sendromun patofizyolojisi henüz tam olarak aydınlatılamamıştır. Bu konuda yapılan çalışmalarda büyüme hormonu, insülin, grelin, leptin düzeyindeki değişiklikler olduğu gösterilmiştir(26). İnfantlarda nedeni açıklanamayan büyüme ve gelişme geriliğinde ayırıcı tanıda hipotalamik bölgede yer işgal eden lezyonlar mutlaka akla getirilmelidir.
Optik sinirin tutulduğu tümörlerde okulda görme problemleri, monooküler görme kaybı görülmektedir. Kiazmatik lezyonlarda özellikle görme keskinliğinde azalma beklenir. Kiazmadan sonraki optik yolda ve oksipital korteks lezyonlarında hemianopsiler, marcus gunn pupil (afferent pupil defekti), nistagmus görülebilmektedir (5).
Tablo 2.9. Çocuklarda SSS Tümörlerinin Yerleşim Yerlerine göre Prezentasyonu
Supratentorial tümörler • Kafa içi basınç artışına ait
bulgular* (%47) • Nöbet (%38)
• Fokal nörolojik bulgular (%17)
• Papilödem *(%21)
• Baş ağrısı* (%11) • Hemipleji (%10) • Bulantı kusma *(%8) • Makrosefali* (%6)
Posterior fossa tümörleri • Bulantı ve kusma* (%75) • Başağrısı* (%67) • Anormal yürüyüş ve koordinasyon bozukluğu (%60) • Papilödem* (%34) • Anormal göz hareketleri (%20) • Letarji (%13)
• Kusma olmadan bulantı* (%10) • Kaza ile kafa travması (%5)
• Kafa içi basınç artışına ait bulgular* (%9)
• Kilo kaybı (%9)
• Fokal motor güçsüzlük (%9) • Makrosefali* (%7)
• Bilinç bozukluğu (%7) • Vertigo ve işitme ile ilgili
semptomlar (%7) • Şaşılık (%6) • Ense sertliği (%6) • Baş eğme Beyinsapı tümörleri • Anormal yürüyüş ve koordinasyon bozukluğu (%78) • Kranial sinir paralizisi (%52) • Piramidal bulgular (%33) • Baş ağrısı* (%23) • Fokal motor güçsüzlük (%19) • Şaşılık (%19) • Yüz felci (%15) • Papilödem* (%13)
• Kafa içi basınç artışına ait bulgular* (%10)
• Anormal göz hareketleri (%6) • Davranış değişiklikleri ve okul
başarısında düşüklük (%5)
Orta hat tümörleri • Baş ağrısı* (%49)
• Anormal göz hareketleri ve şaşılık (%21)
• Bulantı ve kusma* (%19) • Papilödem* (%18)
• Görme keskinliğinde azalma (%16)
• Kafa içi basınç artışına ait bulgular* (%13)
• Diabetes insipitus (%12)
• Anormal yürüyüş ve koordinasyon bozukluğu (%10)
• Davranış değişiklikleri ve okul başarısında düşüklük (%9)
• Bilinç düzeyinde değişiklik (%9) • Optik atrofi (%9)
• Görme alanında azalma (%8) • Nöbet (%7)
• Hemipleji (%7)
• Fokal motor defisit (%7) • Gelişim geriliği (%7) • Boy kısalığı (%7) • Kilo kaybı (%5)
• Vertigo ve işitme ile ilgili semptomlar (%5)
Spinal kord tümörleri • Sırt ağrısı (%67) • Anormal yürüyüş ve koordinasyon bozukluğu (%42) • Spinal deformite (%39) • Fokal motor güçsüzlük (%21) • Sfinkter disfonksiyonu (%20) • Üst ekstremite hareketlerinde azalma (%17) • Gelişim geriliği (%8) • Baş eğme (%7) • Baş ağrısı* (%7)
* İntrakranial basınç artışına bağlı semptom ve bulgular
Reproduced with permission from: Wilne, S, Collier, J, Kennedy, C. Presentation of childhood CNS tumours: a systematic review and meta-analysis. Lancet Oncol 2007; 8:685. Illustration used with the permission of Elsevier Inc. All rights reserved.
Pineal bölge tümörlerinde yukarı bakış paralizisi, akomodasyona reaktif olan fakat ışığa reaktif olmayan pupiller dilatasyon, konverjansla nistagmus görülmektedir ve durum parinaud sendromu olarak tanımlanmaktadır (5).
SSS tümörleri basit, kompleks, parsiyel veya jeneralize nöbetler ile prezente olabilmektedir. Yapılan bazı çalışmalarda nöbet ile başvuran hastalarda kafa içi basınç artışı bulguları ile başvuran hastalara göre sonuçların daha iyi olduğu saptanmıştır (27).
Tümör nedenli kafa içi basınç artışına bağlı baş ağrısının karakteri migren ya da gerilim tipi baş ağrısından farklıdır. Tümör ile ilişkili baş ağrısı genelde gece uykudan uyandıran, sabah ilk uyanma sırasında mevcut olan ve ayağa kalkma ile hafifleyen, kusma ile hafifleyen, valsalva manevrası ile artış gösteren, daha çok oksipital ve frontal bölgelerde lokalize olan vasıftadır. Öyküde yakın zamanda ortaya çıkıp devam ettiği ve gün geçtikçe ağrının şiddet ve frekansının artış gösterdiği öğrenilir. Baş ağrısı yakınması ile başvuran hastanın öyküsü mutlaka “kırmızı bayrak” bulguları sorularak derinleştirilmelidir(23,28). Kırmızı bayrak bulguları tablo 2.10’da verilmiştir.
Tablo 2.10. Beyin Tümörü ile İlişkili Baş Ağrısı İçin “Kırmızı Bayrak” Bulguları
• Okul öncesi çocuklarda açıklanamayan baş ağrısı
• 4 haftadan uzun süre devam eden yeni ortaya çıkmış ciddi veya persistan baş ağrısı
• Oksipital bölgede lokalize baş ağrısı • Gece uyandıran baş ağrısı
• Yatınca veya geceleri şiddetlenen baş ağrısı • Bilinç seviyesinde değişiklik olması
• Anormal baş pozisyonunun (tortikollis veya ense sertliği gibi) eşlik etmesi • Özellikle sabahları olan kusmanın eşlik etmesi
• Daha önceden tanı konmuş baş ağrısının karakterinde değişiklik olması
Şekil 2.2. 0-19 Yaşları Arasında Primer Beyin ve SSS Tümörlerinin Yerleşim Yerlerine göre
Dağılımı, CBTRUS 2007-2011
2.2.5. Santral Sinir Sistemi (SSS) Tümörlerinde Tanı Yöntemleri
Çocukluk çağı beyin tümörleri, tümör yerleşimyeri, tümör biyolojisi ve çocuğun yaşına bağlı olarak çok farklı klinik bulgularla karşımıza geldiği için semptomların başlamasından tanıya kadar geçen süre oldukça değişkenlik göstermektedir. Yapılan çalışmalarda bu sürenin 2,5 ve 3,5 ay arasında değiştiği gösterilmiştir (29). Bu süre mortalite ve morbidite ile doğrudan ilişkili olduğu için beyin tümöründen şüphelenilen bir hastanın değerlendirilmesinin acil olduğunu bilerek hareket etmemiz gerekmektedir.
Olfaktor tümörler 0% Serebellum 16% Pitiuter ve kraniofarengeal kanal 14% Diğer; beyin 14% Beyin sapı 10% Temporal lob 7% Frontal lob 6% Kranial sinirler 6% Ventrikül 6% Serebrum 5% Spinal kord ve kauda equina 5% Paryatel lob 3% Pineal bölge 3% Meninks 3%
Diğer; sinir sistemi 2%
Reproduced with permission from: CBTRUS Statistical Report: NPCR and SEER, 2007-2011. Copyright ©2014 Central Brain Tumor Registry of the United States.
Başlangıç değerlendirmesi rutin genel pediatri yaklaşımında olduğu gibi detaylı bir anamnez, tam bir fizik muayene ile başlamaktadır. Ayrıntılı nörolojik değerlendirme ve nörogörüntüleme yöntemleri tanıda önemli diğer basamakları oluşturmaktadır. Bilgisayarlı tomografi (BT) ve manyetik rezonans görüntüleme (MRG) tanı amacı ile en sık başvurulan yöntemlerdir. MRG, beyin tümörleri için BT ile karşılaştırıldığında daha üstün bir görüntüleme yöntemidir. Ancak BT, daha yaygın olarak bulunması, gerekli çalışma süresinin oldukça kısa olması ve genellikle sedasyon gerektirmemesi nedeni ile genellikle tercih edilen ilk görüntüleme yöntemi olmaktadır. BT intrakranial basınç artışı olduğundan şüphelenilen, tıbbi açıdan instabil bir çocuk için acil durumlarda ilk olarak tercih edilmesi gereken tanı yöntemidir. Bununla birlikte, normal bir kranial BT görüntülemesinin bir beyin lezyonu olasılığını tamamen dışlamak için yeterli olmadığını bilmek ve klinik şüphenin devam ettiği olgularda MRG ile bulgularımızı destelemek gerekmektedir.
Tümörün kesin tanısı ve tümör türünün spesifikteşhisi ancak histolojik incele ile mümkün olmaktadır.
2.2.6. Santral Sinir Sistemi (SSS) Tümörlerinin Bir Alt Grubu Olan Atipik Teratoid Rabdoid Tümörün (AT/RT) Genel Özellikleri
Santral sinir sisteminin (SSS) Atipik teratoid rabdoid tümörü (AT/RT) ilk olarak 1987 yılında Rorke ve arkadaşları tarafından tanımlanmıştır (7,30). Çoğunlukla üç yaşından küçük çocuklarda görülen oldukça malign bir tümördür. Tüm santral sinir sistemi tümörlerinin %1-2’sini oluştururken infant yaş grubundaki çocuklarda SSS tümörlerinin %10’unu oluşturmaktadır(4,7). SSS’nin AT/RT’ü oldukça agresif seyirlidir ve prognozu diğer malign beyin tümörleri ile karşılaştırıldığında son derece kötüdür. SSS’nin AT/RT’ü tanısı alan hastalarda 5 yıllık sağ kalım % 39,5 olarak bulunmuştur (4). Bildirilen yaşam süreleri 0,5 ile 11 ay arasında değişmektedir (7,8).
AT/RT nadir bir SSS tümörüdür ancak 0-2 yaş arasındaki çocuklarda medulloblastom ve PNET ile benzer sıklıkta görülmektedir (4). Erişkin yaş grubunda oldukça nadir görülmektedir ve literatürde olgu sunumları olarak karşımıza çıkmaktadır (31–33).
Dünya Sağlık Örgütü (DSÖ) AT/RT’ü ilk olarak 2000 yılında yaptığı sınıflamada çocukluk çağı SSS tümörlerinin bir alt sınıfı olarak tanımlamıştır. 2016 yılında yapılan sınıflamada da yer almaktadır ve AT/RT ile ilişkisi gösterilmiş olan hSNF5/INI1 genindeki değişikliklerin tümör dokusunda gösterilmesi kesin tanı için gerekmektedir(22).
AT/RT, histolojik görünüm olarak oldukça yoğun, son derece artmış selülerite gösteren andiferansiye hücrelerden oluşmaktadır. AT/RT’ün içerdiği rabdoid hücrelerin temel özellikleri, eksantrik yerleşimli çekirdeğe, belirgin çekirdekçiğe, eozinofilik sitoplazmaya ve eozinofilik inklüzyon cisimlerine sahip olmalarıdır. Sıklıkla tümör dokusunda nekroz ve yüksek oranda mitotik aktivite görülmektedir (7,34).
Tümör histolojik olarak rabdoid hücrelerin yanı sıra primitif nöroepitelyal hücreler, mezenkimal hücreler ve epitel dokusu bölgeleri de içerebilmektedir. Tipik rabdoid hücreler genellikle tümörün yalnızca bir bölümünü oluştururken geri kalan doku çeşitli hücre tiplerinden veya önceden var olan bir primer lezyondan oluşur. Rabdoid transformasyon meningiomda, astrositom ve gliomada da bildirilmiştir(35).
AT/RT hücreleri, epitel membran antijeni (EMA), vimentin (VIM), düz kas aktin (SMA) ve glial fibriler asidik protein (GFAP) dahil olmak üzere geniş bir yelpazede epitel, mezenkimal, glial ve sinir belirteçleri ile immunohistokimyasal boyanma gösterirler.AT/RT için en yaygın olarak bilinen tümör süpresör ve tanısal biyolojik belirteç hSNF5/INI1’dir (36).
Histolojik özellikleri nedeni ile sıklıkla primitif nöroektodermal tümör (PNET), medulloblastom, germ hücreli tümör, koroid pleksus karsinomu ile karışabilmektedir (7,30). Histolojik olarak diğer tümörlerle oldukça benzer özellikler taşıması nedeni ile SSS’nin AT/RT’ü olgularının sıklıkla yanlış tanı alabildiği gösterilmiştir (4,8). Anti-hSNF5/INI1antikorutanısal bir araç olarak kullanılmadan önce yanlış tanı oranlarının daha yüksek olduğu bilinmektedir. Bu bulgu, malign pediatrik SSS tümörlerinde hSNF5/INI1protein ekspresyonunu sistematik olarak analiz etmenin akılcılığını vurgulamaktadır (4).
Aday bir tümör supresör gen olarak tanımlanan hSNF5/INI1 ile ilişkisi gösterilmiş olan ilk primer pediatrik beyin tümörü AT/RT’dür (9). SSS, renal
ve ekstrarenal yerleşimli rabdoid tümörlerin büyük çoğunluğunda hSNF5/INI1 geninde genomik değişiklikler olduğu gösterilmiştir (9).
SSS’nin AT/RT'ünü diğer primitif nöroektodermal tümörlerden ayırt etmede kullanılabilecek spesifik klinik özellikler bulunmamaktadır. AT/RT'lerin yaklaşık yarısı, posterior fossa'da yerleşim göstermekle birlikte suprasellar bölge, pineal bölge, omurilik ve ekstramedüller bölgeler de dahil olmak üzere santral sinir sisteminin tüm bölgelerine yerleşebildiği bilinmektedir (37).
Posterior fossada yerleşen tümörler öncelikle serebellopontin açıya yerleşme eğilimi göstermektedir. Supratentorial bölgeye yerleşen tümörler genelde büyük boyutludur ve genelde içlerinde belirgin kistik, nekrotik alanlar içermektedir. Tümörler intraaksiyal veya ekstraaksiyal olabilmektedir. Genellikle komşu yapılardan başlayarak invazyon göstermektedir (37).
Başlıca otopsi materyallerinin incelendiği çalışmalarda veya retrospektif nöropatolojik incelemelere dayanan çalışmalarda elde edilen verilere dayanarak hastaların % 35-40'ında tanı sırasında leptomeningeal tutulumun mevcut olduğu düşünülmektedir (7).
SSS’nin AT/RT’ünün bilgisayarlı tomografide (BT) görünümü tipik olarak hiperdens bir kitle şeklindedir. Nadiren kalsifikasyon içermektedir. Kistler supratentorial alanda yaygındır, posterior fossada daha az görülmektedir. T1 ağırlıklı manyetik rezonans görüntülemede (MRG) tümör kitlesi, tipik olarak, intratümöral kanamaya sekonder oluşan hemoraji odakları ile izointens görünmektedir. T2 ağırlıklı sekanslarda tümörün görünümü, tümör selüleritesi, kanamalı, nekrotik ve kistik komponentlerin birleşimine bağlı olarak heterojen (hipo-, izo- ve hiperintens odakların karışımı şeklinde) olabilmektedir(37) .
AT/RT’lü olgularının çoğunlukla çok küçük yaşta olması, tümör yerleşiminin beyin sapında, kranial sinirlere çok yakın yerleşmesine bağlı olarak cerrahi yaklaşımınınzor olması ve hastalığın son dereceagresif seyirli olması nedeni ile tedavi seçenekleri oldukça kısıtlıdır. Tedavide kullanılan ortak bir tedavi protokolü oluşturulamamıştır. Nadir görülen bir tümör olması ve sağ kalım süresinin kısa olması nedeni ile uygulanan tedavi yöntemlerinin etkinliğini karşılaştırmak için yeterli prospektif çalışma yoktur. Bu nedenle bu konu ile ilgili bilgilerimiz kısıtlıdır ve kapsamlı çalışmalara ihtiyaç duyulmaktadır.
Tanı anında hastalar çok küçük yaşta genelde infant yaş grubunda oldukları için cerrahi sonrası adjuvan tedavi olarak radyoterapi tercih edilememektedir. Daha çok kemoterapi ile ilgili deneyimler mevcuttur. Yüksek doz siklofosfamid, sisplatin, etoposid ve vinkristin kombinasyonu veya ifosfamid, karboplatin, vinkristin ve etoposid kombinasyonunun kullanıldığı tedavi rejimlerine yanıt alındığı bildirilmiştir. Bununla birlikte, farklı kliniklerde farklı kemoterapi protokolleri uygulandığı için ve tek bir rejim alan hasta sayısı oldukça az olduğu için kemoterapi verilerini değerlendirmek de son derece zordur. Cerrahi ve kemoterapialan çocuklar için genel sağ kalım oranlarının tanıdan sonraki 12 ayda% 20 veya daha az olduğu bulunmuştur(37,38).
2.3. Kanser ve Genetik
2.3.1.Kanser Genetiğinde Temel Kavramlar
Somatik mutasyon: Germ hücreleri dışındaki vücut hücrelerinde, yani somatik hücrelerde ortaya çıkan mutasyonlar somatik mutasyon olarak tanımlanmaktadır. Bu tür mutasyonlar sonraki kuşağa aktarılmadığı için biyolojik sonuçları yalnızca ortaya çıktıkları bireyi etkilemektedir. Kalıtsal özellik göstermeyen, sporadik kanserlerin gelişiminde somatik mutasyonlar rol oynamaktadır. Kanserin somatik genetik değişikliklerden kaynaklandığı ilk olarak Burkit lenfoma üzerinde yapılan çalışmalarda gösterilmiştir(39).
Germline mutasyon: Gonadlardaki germ hücrelerinde (eşey hücresi; sperm ya da ovdum) ortaya çıkan mutasyonlar germline mutasyon olarak tanımlanmaktadır. Bu tür bir mutasyon taşıyan bireyler bunu çocuklarına aktarabilirler. Mutasyonu alan çocuk yalnızca germ hücrelerinde değil, vücudunun tüm hücrelerinde bu mutasyonu taşıyacaktır. Kalıtsal kanserlerden germline mutasyonlar sorumlu tutulmaktadır(39).
Protoonkogen: Hücre çoğalması normalde fizyolojik gereksinimlere göre ve kontrollü olarak yürütülmektedir. Protoonkogenlerhücre