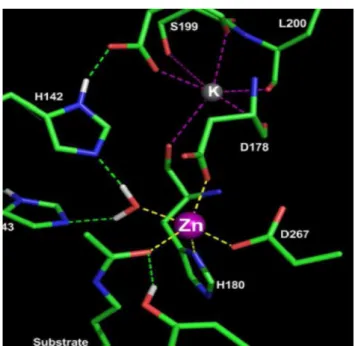
Docking study of resveratrol like molecules on histone deacetylase
Tam metin
Şekil
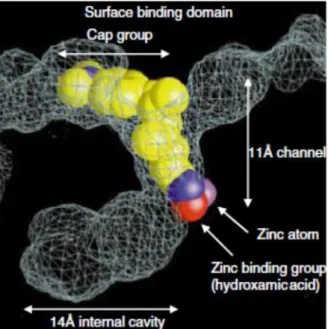
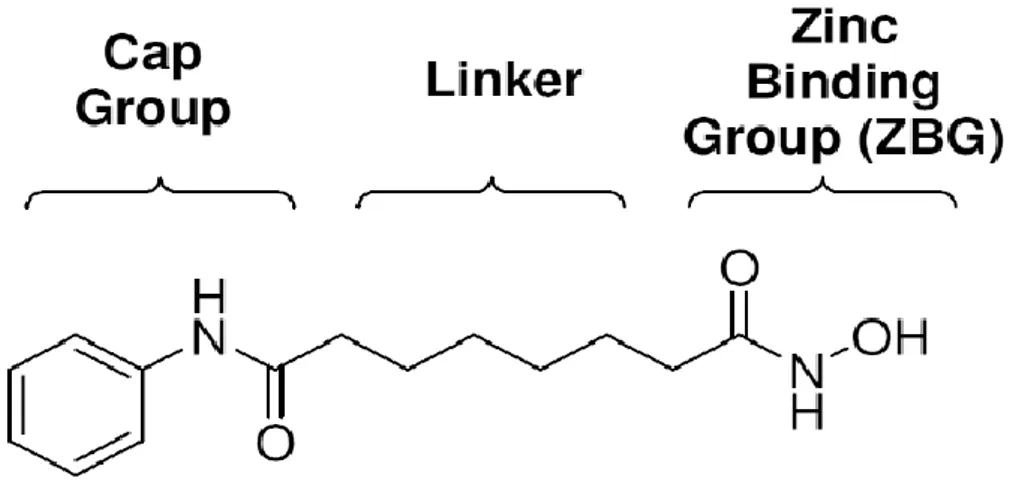
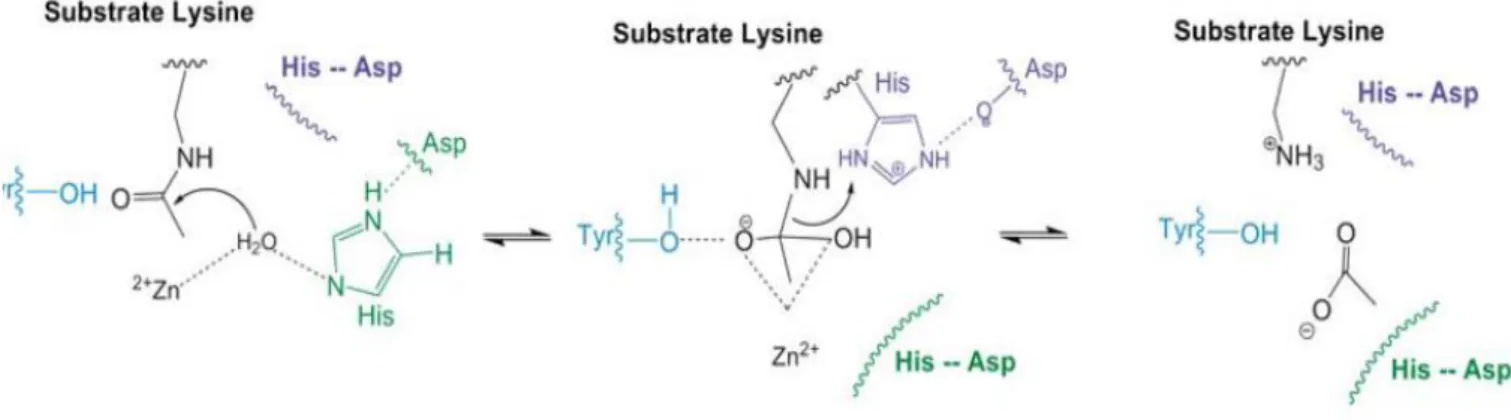
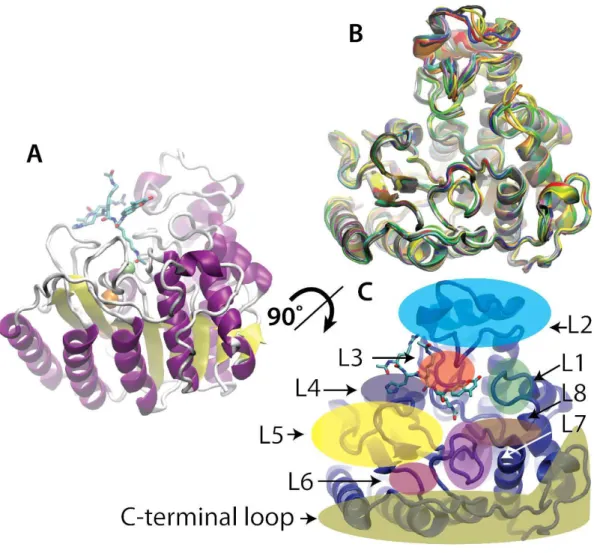

Benzer Belgeler
These compounds have been shown to possess promising histone deacetylase inhibition activities via in vitro fluorometric assay and molecular docking studies.. Key words:
In all 3 graphs (Figures 1a-1c), the bars on the negative side indicate that the compound either failed to dock to the enzyme or was bound very poorly. It is clear that the AutoDock
Synthesis, Biological Evaluation, and Computer-Aided Drug Designing of New Derivatives of Hyperactive Suberoylanilide Hydroxamic Acid Histone Deacetylase Inhibitors. Chem Biol
Analytical methods are classified according to the measurement of some quantities proportional to the quantity of analyte. Classical Methods and
Potentiometry is a quantitative analysis of ions in the solution using measured potentials in an electrochemical cell formed with a reference electrode and a suitable
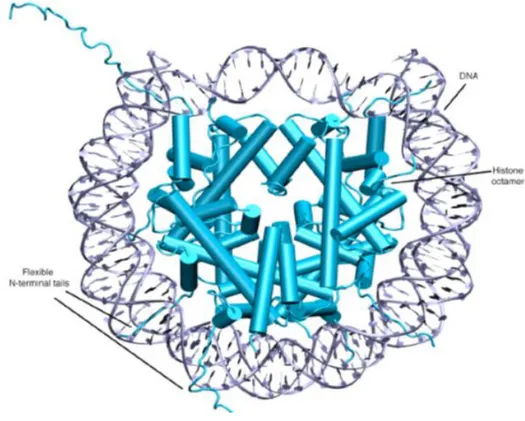
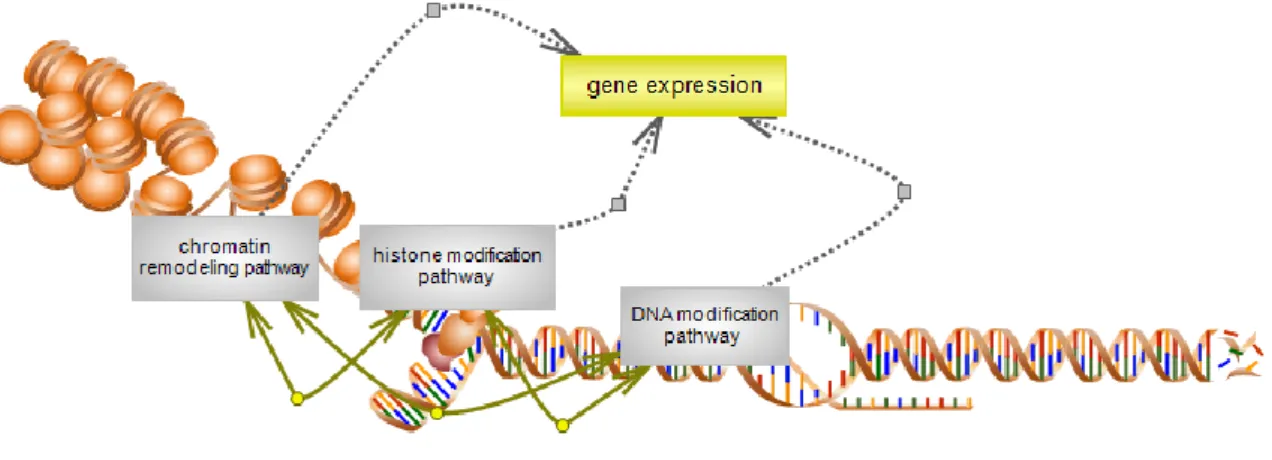
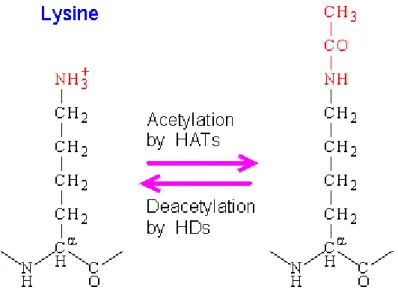
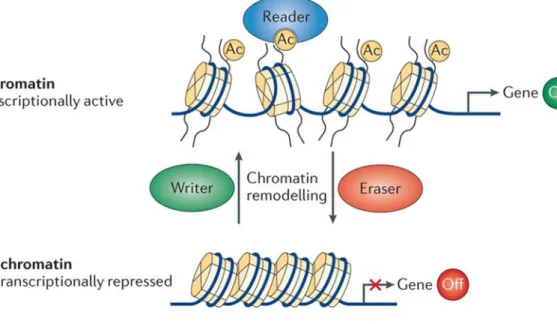
• In interphase (nondividing) cells, most of the chromatin (called euchromatin) is relatively decondensed and distributed. • During this period of the
Boltzmann disribution law states that the probability of finding the molecule in a particular energy state varies exponentially as the energy divided by k
When considering women empowerment, indicators in this thesis such as gender role attitude of women and controlling behavior of husbands, personal and relational
