T.C.
EGE ÜNİVERSİTESİ TIP FAKÜLTESİ
TIBBİ BİYOKİMYA ANABİLİM DALI
DİYABETİK NEFROPATİDE FİBROBLAST BÜYÜME FAKTÖRÜ-23
(FGF-23)
TIPTA UZMANLIK EĞİTİMİ TEZİ
Dr. Eda ALKAYA
DANIŞMAN
Prof. Dr. Sara HABİF
İZMİR
OCAK 2014
Bu çalışma EÜTF Bilimsel Araştırma Projeleri Alt Komisyonu tarafından desteklenmiştir. Proje No: 2012-TIP-001
i
TEŞEKKÜR
Ege Üniversitesi Tıp Fakültesi Tıbbi Biyokimya Anabilim Dalı ve Klinik Biyokimya Bilim Dalı’ndaki uzmanlık eğitimim süresince tecrübe, bilgi ve emeğini hoşgörü ile paylaşan ve bana her konuda destek olan değerli hocam ve tez danışmanım Prof. Dr. Sara Habif’e,
Bilgi, deneyim ve desteklerini özveriyle aktaran değerli hocalarım Prof. Dr. Eser Sözmen’e, Prof. Dr. Taner Onat’a, Prof. Dr. Dilek Özmen’e, Prof. Dr. Işıl Mutaf’a, Prof. Dr. Nevbahar Turgan Temeltaş’a, Prof. Dr. Ferhan Sağın’a, Prof. Dr. Gülinnaz Ercan’a, Prof. Dr. Hikmet Hakan Aydın’a, Prof. Dr. Yasemin Akçay’a, Prof. Dr. Zuhal Parıldar’a, Prof. Dr. Handan Ak’a, Prof. Dr. Ceyda Kabaroğlu’na, Doç. Dr. Burcu Barutçuoğlu’na, Doç. Dr. Güneş Başol’a, Doç. Dr. Ali Mert Özgönül’e ve Doç. Dr. Ebru Sezer’e, ayrıca şu an emekli olan değerli hocam Prof. Dr. H. Oya Bayındır’a,
EÜTF İç Hastalıkları Anabilim Dalı, Geriatri Bilim Dalı’ndan Doç. Dr. Zeliha Fulden Saraç’a ve EÜTF İç Hastalıkları Anabilim Dalı, Nefroloji Bilim Dalı’ndan Doç. Dr. Meltem Seziş Demirci’ye,
Tezimin laboratuar çalışmaları sırasında büyük yardımlarını gördüğüm Y. Kimya Müh. Serap Güven Top’a, Biyolog L. Nurten Çetiner’e ve Biyolog Merve Kurşunel’e,
Uzmanlık eğitimim boyunca beraber çalıştığım uzman, asistan ve teknisyen arkadaşlarıma ve diğer çalışanlara,
Her zaman yanımda olan aileme,
En içten sevgi, saygı ve teşekkürlerimi sunarım.
ii
ÖZET
Diabetes mellitus, komplikasyonları nedeniyle yaşam kalitesi ve süresini olumsuz yönde etkileyen kronik bir hastalıktır. 2030 yılına kadar hasta sayısının 439 milyona yükseleceği öngörülmekte olup, diyabete bağlı komplikasyonların ortaya çıkışını engellemek ya da geciktirmek büyük bir önem taşımaktadır.
Fibroblast Büyüme Faktörü-23 (FGF-23), fosfor metabolizmasındaki düzenleyici rolü nedeniyle, bir fosfatoninin olarak tanımlanmakta olup, kronik böbrek hastalarında, böbrek işlevindeki azalma ile birlikte artış gösterdiğine ve bu artışı serum kalsiyum, fosfor, parathormon düzeylerinde bir değişiklik olmadan önce ortaya çıktığına ilişkin çalışmalar bulunmaktadır.
Bu ön bilgilerin ışığı altında, bu çalışmada, tip 2 diyabetik hastalarda, FGF-23’ün nefropati gelişimindeki rolünün araştırılması, diyabetik nefropati gelişmiş ve gelişmemiş olgularda saptanan değişikliklerin karşılaştırılması ve FGF-23’ün böbrek hasarı gelişiminin erken bir göstergesi olarak kullanılıp kullanılamayacağının araştırılması amaçlandı.
Çalışma grubu, diyabetik nefropati varlığına göre iki temel gruptan oluşturuldu (Grup 1: nefropatisiz, n=60, Grup 2: nefropatili, n=60). Nefropatili olgular da nefropatinin derecesine göre 2 alt gruba ayrıldı (Grup 2a: mikroalbuminüri, n=36, Grup 2b: makroalbuminüri, n=24). Olgularda kan ve spot idrardaki rutin parametreler yanı sıra, FGF-23, 25(OH)D vitamini, 1,25(OH)2D vitamini, PTH düzeylerinin gruplar arasındaki farklılıkları ve parametreler
arasındaki ilişki araştırıldı.
1,25(OH)2D vitamini, Grup 2’de Grup 1’e kıyasla anlamlı derecede düşük idi (sırasıyla 19.7 ±
5.4 pg/mL ve 22.0 ± 9.3 pg/mL, p=0.009). FGF-23 ve PTH düzeyleri Grup 2’de Grup 1’e kıyasla anlamlı düzeyde yüksek idi (sırasıyla FGF-23: 16.2 ± 19.9 pg/mL ve 6.4 ± 6.4 pg/mL, p<0.001; PTH: 83.9 ± 125.6 pg/mL ve 60.7 ± 61.6 pg/mL, p=0.001). FGF-23 ile albuminüri derecesi, 1,25(OH)2D vitamini, HbA1c ve HDL-Kolesterol arasında, ayrıca
makroalbüminürili olgularda, eGFR ve FGF-23 arasında anlamlı korelasyon mevcuttu.
Bu çalışmada elde edilen veriler, diyabetik nefropati patogenezinde FGF-23’ün rolünü desteklemektedir. Diyabette fosfor dengesinin düzenlenmesinde bir belirteç olarak FGF-23’ün kullanımı ile ilgili olarak daha geniş kapsamlı çalışmalara gereksinim vardır.
iii
ABSTRACT
Diabetes mellitus is a chronic disease which affects the quality and time of life due to its complications. It is estimated that the number of patients will increase up to 439 million till 2030. It is too important to prevent and delay the occurrence of diabetic complications.
The Fibroblast Growth Factor-23 (FGF-23) is defined as a phosphatonin due to its regulatory role in the phosphorus metabolism. There are some studies introduce that, FGF-23 levels increased with the functional deterioration in chronic kidney patients which occurred before a change in serum calcium, phosphorus, parathormone levels.
In the light of this introductory information, the aim of this study was to investigate the role of FGF-23 in diabetic nephropathy, to comparate the FGF-23 and related data in cases with and without nephropathy and to investigate whether FGF-23 could be used as an early indicator in the development of renal damage in type 2 diabetic patients.
The work group was consisted of two main groups which were designed according to the presence of diabetic nephropathy (Group 1: without nephropathy, n=60, Group 2: with nephropathy, n=60). The nephropathy cases were also divided into two subgroups (Group 2a: microalbuminuria, n= 36, Group 2b: macroalbuminuria, n=24). Besides the routine parameters in blood and spot urine samples, differences and the correlations were investigated for FGF-23, 25(OH)D vitamin, 1,25(OH)2D vitamin and PTH between groups.
1,25(OH)2D vitamin was significantly low in Group 2 compared to Group 1 (respectively
19.7 ± 5.4 pg/mL and 22.0 ± 9.3 pg/mL, p=0.009). FGF-23 and PTH levels were higher in Group 2 compared to Group 1 (respectively FGF-23: 16.2 ± 19.9 pg/mL and 6.4±6.4 pg/mL, p<0.001; PTH: 83.9 ± 125.6 pg/mL and 60.7 ± 61.6 pg/mL, p=0.001). There were a significant correlation between FGF-23 and albuminuria, 1,25(OH)2D vitamin, HbA1c,
HDL-Cholesterol and also in the macroalbuminuric patients between eGFR and FGF-23.
The data obtained data in this study is supporting the role of FGF-23 in the pathogenesis of diabetic nephropathy. More comprehensive studies are needed, relating to the use of FGF-23 as an indicator in the regulation the phosphorus balance in diabetes.
iv İÇİNDEKİLER Sayfa TEŞEKKÜR ……… i ÖZET ………... ii ABSTRACT ……… iii İÇİNDEKİLER ………... iv KISALTMALAR ……… vii ŞEKİLLER ……….. ix TABLOLAR ………... xi 1. GİRİŞ VE AMAÇ ……… 1 2. GENEL BİLGİLER ………. 3 2.1. Diabetes Mellitus ………... 3 2.1.1. Tanım ………... 3 2.1.2. Epidemiyoloji ……….. 3 2.1.3. Sınıflandırma ………... 4 2.1.4. Tanı ……….. 4
2.1.5. Tip 1 Diabetes Mellitus ……….……….. 7
2.1.6. Tip 2 Diabetes Mellitus ……….……….. 8
2.1.7. Diabetes Mellitus’un Komplikasyonları ……….. 10
2.1.8. Diyabetik Nefropati ………. 11
2.2. Kalsiyum - Fosfor Metabolizması ………. 19
2.2.1. Kalsiyum ……….. 19
2.2.2. Fosfor ………... 23
2.2.3. Parathormon ………. 26
v
2.2.5. Kronik Böbrek Hastalığında Kalsiyum - Fosfor Metabolizması …………. 30
2.3. Fibroblast Büyüme Faktörü-23 (FGF-23) ………. 32
2.3.1. FGF-23’ün Yapısı ……… 32
2.3.2. FGF Reseptörleri ………. 33
2.3.3. Klotho Proteini ……… 35
2.3.4. FGF-23’ün Fizyolojik Etkileri ………. 35
2.3.5. FGF-23 Seviyelerinin Düzenlenmesi ……….. 37
2.3.6. Fosfor Homeostazında FGF-23’ün Rolü ………. 39
2.3.7. FGF-23 ile Kalıtsal İlişkili Hastalıklar ………..….. 40
2.3.8. Onkojenik Osteomalazi……… 42
2.3.8. Kronik Böbrek Hastalığı ve FGF-23 ………... 42
3. ARAÇ, GEREÇ VE YÖNTEMLER ………... 47
3.1. Çalışma Gruplarının Oluşturulması ………... 47
3.2. Örneklerin Alınması, Saklanması, Hazırlanması ………... 48
3.3. Cihazlar ve Sarf Malzemeleri ……… 48
3.4. 25(OH)D Vitamini Ölçümü ………... 49
3.5. 1,25(OH)2D Vitamini Ölçümü ………... 50
3.6. Parathormon Ölçümü ………. 50
3.7. FGF-23 Ölçümü ………. 50
3.8 Tam Kan Sayımı Ölçümü ………... 52
3.9. Rutin Biyokimyasal Testlerin Ölçümü ……….. 52
3.10. Fraksiyone Fosfor Atılımının Hesaplanması ………... 54
3.11. Glomerüler Filtrasyon Hızının Hesaplanması ………. 54
3.12. İstatistiksel Analiz ………... 54
4. BULGULAR ………... 55
vi
6. KAYNAKLAR ………... 88
7. EKLER
EK 1: Bilgilendirilmiş Gönüllü Olur Formu EK 2: Olgu Rapor Formu
vii
KISALTMALAR
1,25(OH)2D : 1,25-dihidroksivitamin D
25(OH)2D : 25-dihidroksivitamin D
ADA : Amerikan Diyabet Birliği
ADHR : Otozomal dominant hipofosfatemik raşitizm
AGE : İleri glikasyon son ürünleri
ASARM : Asidik serin-aspartat zengin motif
BÇ : Bel çevresi
BMI : Vücut kütle indeksi
BMP1 : Kemik morfogenetik protein 1
Ca : Kalsiyum
CaSR : Kalsiyum duyarlı reseptör
CRP : C- Reaktif protein
CTGF : Bağ Dokusu büyüme faktörü
DCCT : Diyabet Kontrolü ve Komplikasyonları Çalışması
DM : Diabetes Mellitus
DMP1 : Dentin matriks asidik fosfoprotein 1
DN : Diyabetik nefropati
ECM : Ekstrasellüler matriks
FeP : Fraksiyone fosfor atılımı
FGF : Fibroblast büyüme faktörü
FGFR : Fibroblast büyüme faktörü reseptörü
GAD : Glutamik asid dekarboksilaz
GALNT3 : UDP-N-asetil-α-D-galaktozamin: polipeptid N-asetilgalaktozaminil transferaz 3
GBM : Glomerüler bazal membran
GFR : Glomerüler filtrasyon hızı
GH : Büyüme hormonu
GNAS1 : Guanin nükleotid bağlayıcı protein alfa uyarıcı gen
HbA1c : Hemoglobin A1c
hsCRP : Yüksek duyarlıklı C-Reaktif protein HSPG : Heparan sülfat proteoglikan zinciri
viii
ICA : Adacık hücre antikorları
IFG : Bozulmuş açlık glukozu
IGF : İnsülin benzeri büyüme faktörü
IGT : Bozulmuş glukoz toleransı
KBH : Kronik böbrek hastalığı
KÇ : Kalça çevresi
MAPK : Mitojen ile aktive olan protein kinaz
MAU : Mikroalbumin
MEPE : Matriks ekstraselüler fosfoglikoprotein NADPH : Nikotinamid Adenin Dinükleotid Fosfat
NGSP : Ulusal Glikohemoglobin Standardizasyon Programı
OGTT : Oral glukoz tolerans testi
PHEX : X kromozomu üzerindeki endopeptidaz benzeri fosfat düzenleyici gen
Pi : İnorganik fosfor
PPAR-γ : Peroksizom proliferatör-aktive reseptör gama
PTH : Parathormon
PTHrP : Parathormon ilişkili peptid
RANK : Reseptör aktivatör nükleer kappa B RANKL : Reseptör aktivatör nükleer kappa B ligand
ROS : Reaktif oksijen türleri
SDBY : Son dönem böbrek yetmezliği
sFRP4 : Secreted frizzled related protein-4 TGF-β : Transforme edici büyüme faktörü beta
TNF-α : Tümör nekroz faktör-α
TRPV : Transient reseptör potansiyelli-vanilloid iyon kanalı
u-KIM1 : Kidney injury molecule 1
u-NGAL : Neutrophil gelatinase-associated lipocalin
VDR : Vitamin D reseptörü
VDRE : Vitamin D cevap elementi
VEGF : Vasküler endotelyal büyüme faktörü VLDL : Çok düşük dansiteli lipoprotein
ix
ŞEKİLLER
Şekil Sayfa
2.1. Diyabetik nefropati patogenezi 14
2.2. Diyabetik nefropatinin doğal seyri 16
2.3. İntestinal ve renal tubüler epitel hücrelerinde kalsiyum transportu 21 2.4. Kalsiyum ve fosfat dengesinin PTH ve 1,25(OH)2D ile düzenlenmesi 23
2.5. Renal proksimal tubül epitelinde NaPi taşıyıcı proteinleri aracılığı ile fosfor geri emilimi
25
2.6. Kronik böbrek hastalığında kemik, fosfor ve kalsiyum anormalliklerinin gelişim şeması
32
2.7. FGF-23’ün yapısı ve proteolitik yıkım ile inaktif fragmanların oluşumu 33
2.8. FGF reseptörlerinin yapısı 34
2.9. FGF-FGFR kompleksinin yapısı 34
2.10. Proksimal tubül hücrelerinde FGF-23’ün etkileri 36 2.11. Paratiroid hücresinde FGF-23’ün etkileri 37 2.12. FGF-23 ekspresyonunu düzenleyen sistemik ve kemik kaynaklı faktörler 38
2.13. Fosfat homeostazında kemik-böbrek-paratiroid aksı 40
2.14. Erken evre KBH’da FGF-23’ün rolü 43
2.15. KBH’da Klotho proteini, FGF-23, PTH, 1,25(OH)2D ve fosfat
seviyelerindeki değişiklikler
44
2.16. Son dönem böbrek hastalığında FGF-23’ün rolü 45
3.1. Serum FGF-23 ELISA standart eğrisi 52
4.1. Nefropatisiz (Grup 1) ve nefropatili (Grup 2) olgularda fraksiyone fosfor atılımı değerleri
62
4.2. Nefropatisiz (Grup 1) ve nefropatili (Grup 2) olgularda 25(OH)D vitamini düzeyleri
x
4.3. Nefropatisiz (Grup 1) ve nefropatili (Grup 2) olgularda 1,25(OH)2D
vitamini düzeyleri
63
4.3. Nefropatisiz (Grup 1) ve nefropatili (Grup 2) olgularda PTH düzeyleri 64
4.4. Nefropatisiz (Grup 1) ve nefropatili (Grup 2) olgularda FGF-23 düzeyleri 64
4.6. Diyabetik nefropati evresi ile FeP arasındaki korelasyon grafiği 67
4.7. Diyabetik nefropati evresi ile 1,25(OH)2 D vitamini arasındaki korelasyon
grafiği
68
4.8. Diyabetik nefropati evresi ile PTH arasındaki korelasyon grafiği 68 4.9. Diyabetik nefropati evresi ile FGF-23 arasındaki korelasyon grafiği 69
4.10. FGF-23 ile HbA1c arasındaki korelasyon grafiği 69
4.11. FGF-23 ile HDL-K arasındaki korelasyon grafiği 70
4.12. FGF-23 ile kalsiyum arasındaki korelasyon grafiği 70
4.13. FGF-23 ile fosfor arasındaki korelasyon grafiği 71
4.14. FGF-23 ile FeP arasındaki korelasyon grafiği 71
4.15. FGF-23 ile PTH arasındaki korelasyon grafiği 72
4.16. FGF-23 ile 1,25(OH)2D arasındaki korelasyon grafiği 72
4.17. İleri evre nefropatili (Grup 2b) olgularda FGF-23 ile eGFR arasındaki korelasyon grafiği
xi
TABLOLAR
Tablo Sayfa
2.1. Diabetes Mellitus’un etiyolojik sınıflandırması 5
2.2. Diyabet tanı kriterleri 6
2.3. Artmış diyabet riski kategorileri 7
2.4. Albumin atılım hızlarının tanımı 16
2.5. Diyabetik nefropati evreleri 18
4.1. Grup 1 ve Grup 2’de demografik veriler 57
4.2. Grup 1 ve Grup 2 de rutin biyokimyasal parametreler 59
4.3. Grup 1 ve Grup 2’de böbrek işlevlerine ilişkin parametreler 61
4.4. Grup 1 ve Grup 2’de kalsiyum, fosfor metabolizmasını düzenleyen hormon düzeylerine ilişkin parametreler
62
4.5. Grup 1, Grup 2a ve Grup 2b’de kalsiyum, fosfor ve ilişkili hormon parametreleri
1. GİRİŞ VE AMAÇ
Diabetes Mellitus (DM), ülkemizde ve dünyada sıklığı hızla artan ve ciddi komplikasyonları nedeniyle yaşam kalitesini, süresini olumsuz yönde etkileyen kronik bir hastalıktır. Dünya Sağlık Örgütü (WHO) 2000 yılı verilerine göre, dünya genelinde 171 milyon DM hastası bulunmaktadır (1). Diyabetik hasta sayısının, 2010 yılında 285 milyona ulaştığı, bu rakamın 2030 yılına kadar 439 milyona yükseleceği öngörülmektedir (2).
DM sıklığındaki artış ve diyabetik hastaların yaşam süresindeki uzamaya bağlı olarak, diyabetin en önemli komplikasyonlarından biri olan diyabetik nefropati sıklığı da giderek artmaktadır (3). Diyabetik nefropati, tüm dünyada son dönem böbrek yetmezliği (SDBY) nedenleri arasında ilk sırada yer almakta olup, diyaliz uygulanan hastaların büyük bir bölümünü de diyabetik hastalar oluşturmaktadır (3, 4). Tip 2 diyabetli hastalarda, tanıdan yaklaşık 10 yıl sonra mikroalbuminüri prevalansı % 25-40 olarak bildirilmiştir. Klinik nefropati gelişen olguların ise %20’sinde, 20 yıl içinde SDBY gelişmekte olduğu belirtilmektedir (5, 6). Bu nedenle diyabetik nefropati gelişiminin erken dönemde saptanabilmesi, SDBY gelişiminin engellenmesi açısından önemlidir.
Fibroblast büyüme faktörü-23 (FGF-23), FGF ailesinin yakın geçmişte tanımlanmış bir üyesi olup, fosfor metabolizmasında düzenleyici role sahip bir hormondur. FGF-23, hiperfosfatemiye yanıt olarak osteositlerden salgılanmakta; böbreklerden fosfor atılımını arttırmakta, 1,25 dihidroksi D vitamini (1,25(OH)2D) sentezini ve parathormon (PTH)
salgılanmasını baskılamaktadır. Kronik böbrek hastalığı gelişiminde glomerüler filtrasyon hızının (GFR) azalması ve fosfor birikiminin, FGF-23 düzeyini arttırarak, kronik böbrek hastalığının erken evrelerinde fosfor dengesinin korunduğu ve bu şekilde serum fosfor düzeylerinde bir değişiklik saptanmadığı belirtilmektedir (7). Yapılan çalışmalarda da FGF-23 yüksekliğinin, kronik böbrek hastalığının erken evrelerinde hastalığın ilerleyişi ile (8, 9), diyaliz hastalarında ise mortalite ile ilişkili olduğu belirtilmektedir (10, 11).
Kronik böbrek hastalarındaki en sık ölüm nedenini kardiyovasküler hastalıklar oluşturmaktadır (12). Kronik böbrek hastalığında, özellikle de SDBY’de kardiyovasküler risk artışı, vasküler kalsifikasyon ile ilişkilendirilmekte ve özellikle hiperfosfatemi ve PTH düzeylerindeki belirgin artışın vasküler kalsifikasyon gelişimine katkıda bulunduğu belirtilmektedir (13, 14). Bu nedenle kronik böbrek hastalığı etiyolojisindeki en önemli nedenlerden biri olan diyabette de fosfor dengesinin takibi böbrek işlevlerindeki bozulmanın erken dönemde saptanabilmesi açısından önem taşımaktadır.
2
Yapılan kaynak taramasında FGF-23 ile ilgili olarak diyabetik hastalar ile yapılmış az sayıda araştırma bulunmakta olup, ülkemizde böyle bir çalışmaya rastlanmamıştır.
Bu araştırmada, diyabetik hastalarda, fosfor metabolizmasındaki önemi nedeniyle FGF-23’ün, D vitamini ve PTH ile ilişkisinin belirlenmesi, diyabetik nefropati gelişmiş ve gelişmemiş olgularda bu parametrelerde saptanan değişikliklerin karşılaştırılması ve tip II diyabet nedeni ile izlenen hastalarda FGF-23 düzeylerinin fosfor düzeylerinde bir artış ortaya çıkmaksızın böbrek hasarı gelişiminin erken bir göstergesi olarak kullanılıp kullanılamayacağının araştırılması amaçlanmıştır.
3
2. GENEL BİLGİLER
2.1. DİABETES MELLİTUS
2.1.1. Tanım
Diabetes mellitus, insülin sekresyonunda ve/veya insülin etkisindeki kusurlara bağlı olarak organizmanın karbonhidrat, yağ ve proteinlerden yeterince yararlanamadığı, hiperglisemi ile karakterize kronik bir metabolizma hastalığıdır (15). DM ile ilişkili metabolik bozukluklar çeşitli organlarda sekonder patofizyolojik değişiklikler, işlev bozuklukları ve yetmezliklere yol açarak, hastalar yanısıra sağlık sistemi üzerinde de büyük bir yük oluşturmaktadır. A.B.D.’de son dönem böbrek yetmezliği, travmatik olmayan alt ekstremite ampütasyonları ve yetişkin körlüklerinin başlıca nedeninin diyabet olduğu vurgulanmaktadır (16).
2.1.2. Epidemiyoloji
Diyabet görülme sıklığı dünya genelinde dramatik olarak artmaktadır. 1985 yılında 30 milyon olan diyabetik hasta sayısı, 2000 yılında 171 milyona ulaşmıştır (1). Bu rakamın 2030 yılında 439 milyona yükseleceği öngörülmektedir (2). Hem tip 1 hem tip 2 DM prevalansı dünya genelinde artış göstermesine rağmen, endüstrileşme ile birlikte fiziksel aktivitenin azalması, obezitenin yaygınlaşmasına bağlı olarak, tip 2 DM görülme sıklığındaki artış çok daha hızlı olmaktadır (1).
Ülkemizde diyabet sıklığını belirlemeye yönelik olarak, toplum genelini yansıtacak şekilde planlanmış ve uluslararası standartlarda gerçekleştirilmiş ilk çalışma 1997-1998 yıllarında yapılan Türkiye Diyabet, Hipertansiyon, Obezite ve Endokrinolojik Hastalıklar Prevalans Çalışması’dır (TURDEP). Bu çalışmada, toplumda diyabet görülme sıklığı %7 olarak bulunmuş olup, kadınlarda ve kentsel bölgelerde yaşayanlarda oranın daha yüksek olduğu belirlenmiştir (17). Ayrıca diyabet riskinin yaşlanma, obezite, hipertansiyon, ailede diyabet varlığı, eğitimsizlik, gelir düzeyi ve alışkanlıklar ile ilişkili olduğu saptanmıştır (17). 2010 yılında gerçekleştirilen TURDEP II sonuçlarına göre ülkemizde diyabet görülme sıklığı % 13.7’ye ulaşmıştır (18).
4
2.1.3. Sınıflandırma
Günümüzde diyabet sınıflaması, hiperglisemiye neden olan patogenetik süreç temel alınarak yapılmaktadır. Birçok tip 2 diyabetli hastanın yaşamının bir döneminde glisemik kontrolün sağlanması için insülin tedavisine ihtiyaç duyması nedeniyle “insüline bağımlı DM” ve “insüline bağımlı olmayan DM” terimleri terk edilmiştir. Yeni sınıflamadaki diğer bir farklılık, yaşın bir kriter olarak kullanılmamasıdır. Tip 1 DM genellikle 30 yaşından önce ortaya çıkmasına rağmen, otoimmün beta hücre hasarlanması herhangi bir yaşta gelişebilir. 30 yaşın üzerinde DM ortaya çıkan hastalarda ise % 5-10 oranında tip 1 DM saptanmıştır. Tip 2 DM sıklıkla ileri yaşlarda görülmesine rağmen, çocuklar, genç erişkinler ve özellikle obez adölesanlarda görülme sıklığı artmaktadır (16).
Tablo 2.1’de yer alan diyabet sınıflamasında tip1 DM, tip 2 DM, gestasyonel DM ve diğer spesifik tipler olmak üzere 4 ana grup tanımlanmıştır (15). Tip 1 DM tam veya tama yakın insülin eksikliği sonucunda gelişirken, tip 2 DM değişken derecelerde insülin direnci, bozulmuş insülin salınımı ve artmış glukoz üretimi ile karakterizedir. Bozulmuş açlık glukozu veya bozulmuş glukoz toleransı, glukoz hemostazının bozulduğu prediyabetik periyodu tanımlar ve tip 2 DM gelişiminin habercisidirler (16).
2.1.4. Tanı
Diyabet tanı kriterleri 1997 yılında Amerikan Diyabet Birliği (ADA) tarafından açlık plazma glukozunun 126 mg/dL ve üzerinde veya 75 g glukoz ile yapılan oral glukoz tolerans testinde (OGTT) 2. saat plazma glukozunun 200 mg/dL ve üzerinde olması olarak belirlenmiştir. Bu değerlerin saptanmasında temel faktör, retinopati ile plazma glukozu arasında gözlenen ilişki olmuştur. Açlık ve OGTT 2. saat glukoz değerleri ile retinopati incelemesinin yapıldığı üç ayrı kesitsel epidemiyolojik çalışmada retinopati prevelansının lineer olarak artış gösterdiği plazma glukoz değerleri, diyabet tanı eşik değerleri olarak kabul edilmiştir (15, 19).
5
Tablo 2.1. Diabetes Mellitus’un etiyolojik sınıflandırması (15)
I. Tip 1 Diyabet (Genellikle mutlak insülin eksikliğine neden olan β hücre yıkımı vardır.)
A. İdiyopatik (% 10) B. İmmün aracılıklı (% 90)
II. Tip 2 Diyabet (İnsülin direnci zemininde ilerleyici insülin salınım kusuru ile karakterizedir.)
III. Gestasyonel Diyabetes Mellitus IV. Diğer spesifik diyabet tipleri
A. β-hücre fonksiyonlarının genetik defekti 20. Kromozom, HNF-4α (MODY1) 7.Kromozom,Glukokinaz(MODY2) 12. Kromozom, HNF-1α(MODY3) 13. Kromozom, IPF-1 (MODY4) 17. Kromozom, HNF-1β (MODY5) 2. Kromozom, NeuroD1(MODY6) Mitokondriyal DNA
Diğerleri
B. İnsülin etkisindeki genetik defektler Leprekonizm
Lipoatrofik diyabet
Rabson-Medenhall sendromu Tip A insülin direnci
Diğerleri
C. Pankreasın ekzokrin doku hastalıkları Fibrokalkulöz pankreopati Hemokromatoz Kistik fibrozis Neoplazi Pankreatit Travma/pankreatektomi Diğerleri D. Endokrinopatiler Akromegali Aldosteronoma Cushing sendromu Feokromositoma Glukagonoma Hipertiroidi Somatostatinoma Diğerleri
E. İlaç veya kimyasal ajanlar Atipik anti-psikotikler Anti-viral ilaçlar Β-adrenerjik agonistler Diazoksid Fenitoin Glukokortikoidler Α-İnterferon Nikotinik asit Pentamidin Proteaz inhibitörleri Tiyazid grubu diüritikler Tiroid hormonu Vacor Diğerleri F. İnfeksiyonlar Konjenital rubella Sitomegalovirüs Diğerleri
G. İmmun aracılıklı nadir diyabet formları
Anti-insülin reseptör antikorları Stiff-man sendromu
Diğerleri
H. Diyabetle ilişkili genetik sendromlar (Monogenik diyabet formları)
Alström sendromu Down sendromu Friedreich tipi ataksi Huntington korea Klinefelter sendromu Laurence-Moon-Biedl sendromu Miyotonik distrofi Porfiria Prader-Willi sendromu Turner sendromu Wolfram(DIDMOAD) sendromu Diğerleri
6
Mikrovasküler ve daha düşük oranda makrovasküler komplikasyonlar ile korele olması nedeni ile diyabet izleminde kullanılan hemoglobin A1c (HbA1c), ölçüm yöntemindeki standardizasyon eksikliği nedeniyle kısa bir süre öncesine kadar diyabet tanısında yer almamaktaydı. Günümüzde testin standardizasyonu sağlanmış olup, 2010 yılında ADA, Uluslararası Diyabet Uzmanlar Komitesi tarafından % 6.5 eşik değerinin diyabet tanısında kullanılması onaylanmıştır. Bu değer de plazma glukozu gibi retinopati prevalansıyla ilişkilidir. Tanıda kullanılacak HbA1c testi Ulusal Glikohemoglobin Standardizasyon Programı (NGSP) tarafından sertifikalandırılmış bir yöntem ile ölçülmeli ve ölçüm yöntemi Diyabet Kontrolü ve Komplikasyonları Çalışması (DCCT) referans yöntemine göre standardize edilmiş olmalıdır (15, 20).
ADA tarafından 2010 yılında yayınlanan diyabet tanı kriterleri tablo 2.2’de görülmektedir.
Tablo 2.2. Diyabet tanı kriterleri (15)
HbA1c ≥ % 6.5 (NGSP sertifikalı ve DCCT’ye göre standardize yöntem ile ölçüm yapılmalı)*
veya
Açlık plazma gukozu ≥ 126 mg/dL (Kalori alımı en az 8 saat önce kesilmeli)* veya
OGTT’de 2. saat plazma glukozu ≥ 200 mg/dL (OGTT, WHO’nun önerdiği şekilde 75 g glukoz ile yapılmalı)*
veya
Klasik hiperglisemi semptomları veya hipergilisemi krizi olan hastada rastgele plazma glukozu ≥ 200 mg/dL
* Belirgin hiperglisemi yokluğunda 1. 2. ve 3. kriterler test tekrarı ile teyit edilmelidir.
Tanı kriterlerini karşılamayan, ancak normal kabul edilen değerlerin üzerindeki sonuçlar için 1997 ve 2003 yılında Uluslararası Diyabet Uzmanlar Komitesi, “bozulmuş açlık glukozu (IFG)” ve “bozulmuş glukoz toleransı (IGT)” tanımlamalarını yapmıştır. IFG ve IGT diyabet ve kardiyovasküler hastalık gelişimi için önemli risk faktörleridir ve ‘Prediyabet’ olarak olarak kabul edilmektedir (19, 21).
Bozulmuş açlık glukozu ve bozulmuş glukoz toleransı olan kişilerde HbA1c değerleri % 5.5 – 6.0 arasında saptanmaktadır. Prospektif çalışmalarda HbA1c değerleri % 5.5 - 6.0 aralığında olan kişilerde 5 yıllık kümülatif diyabet insidansını % 12 - 25 arasında olduğu belirlenmiştir. HbA1c değerleri % 6.0 - 6.5 olan olgularda ise, diyabet görülme insidansının 10 kat daha
7
yüksek olduğu bildirilmektedir (22-25). Tablo 2.3’te diyabet için yüksek risk kategorileri özetlenmiştir. Bu gruptaki hastaların diyabet, kardiyovasküler hastalık açısından izlem ve koruma programına alınması gerektiği belirtilmektedir (15).
Tablo 2.3. Artmış diyabet riski kategorileri (15)
Açlık plazma glukozu: 100 - 125 mg/dL (IFG)
75g OGTT’de 2. saat plazma glukozu: 140 - 199 mg/dL (IGT)
HbA1c: % 5.7 - 6.4
2.1.5. Tip 1 Diabetes Mellitus
Tip 1 diyabet, mutlak insülin eksikliğine yol açan β hücre yıkımı ile karakterize kronik bir hastalıktır. β hücre hasarlanmasından genetik, çevresel ve immünolojik faktörler sorumludur. Her yaşta görülebilse de, ağırlıklı olarak 30 yaşın altında ortaya çıkar. Çocukluk çağında en sık görülen kronik hastalıklardan biridir (26).
Tip 1 diyabet, immün aracılı ve idiyopatik olarak iki gruba ayrılır. İdiyopatik tip 1 diyabet nadir görülür. Etiyoloji bilinmemekle birlikte kalıtımsaldır, β hücre otoimmünitesine ilişkin immünolojik bulgu yoktur ve HLA ile ilişkili değildir. Hastalık ketoasidoz atakları ile seyreder. Ataklar arasında değişken derecelerde insülin eksikliği görülür (26).
İmmün aracılı tip 1 diyabet, diyabet hastalarının % 5 – 10 kadarını oluşturur (15). Genetik yatkınlığı olan kişilerde doğumda β hücre kitlesi normaldir. Enfeksiyonlar veya çevresel uyaranların otoimmün süreci tetiklemesi sonucunda β hücre yıkımı başlar. β hücre kitlesinin yaklaşık % 80’i hasarlandığında aşikar diyabet ortaya çıkar. Hastaların çoğunda preklinik dönemde immünolojik belirteçler saptanabilir (16).
Tip 1 diyabet riski ile ilişkili 20’den fazla gen saptanmıştır. En güçlü yatkınlık oluşturan gen, 6. kromozomun kısa kolu üzerindeki (6p21.3) HLA bölgesinde lokalize IDDM-1 genidir. HLA kompleksinin polimorfizmi genetik riskin % 40 - 50’sinden sorumludur. Tip 1 diyabete yatkınlık özellikle DQA1*0301, DQB1*0302 ve DQB1*0201 haplotipleri ile ilişkilidir. DRB1*1501 ve DQB1*0602 birçok etnik grupta koruyucu allellerdir (16, 26).
Genetik yatkınlığı olan bireylerde, β hücrelerinde immün toleransın bozulmasına ve otoimmünitenin aktivasyonuna yol açan çevresel faktörlerin başında virüsler, toksinler ve bazı
8
gıda maddeleri gelir. β hücrelerini enfekte eden başlıca virüsler, direkt lizise neden olan ensefalomiyokardit virüsü ile spesifik ya da nonspesifik otoimmiteyi başlatan rubella, retrovirüs, reovirüs tip 1,3, sitomegalovirüs ve kabakulak virüsleridir. Coxsackie B virüsü ise antijenik epitoplarının β hücre yüzeyindeki GAD (glutamik asid dekarboksilaz) molekülü ile benzerliği nedeniyle otoimmüniteyi tetikler. Tip 1 DM gelişimi ile ilişkili diğer çevresel faktörler arasında streptozotosin, aloksan, vakor gibi bakteriyel toksinler, bazı gıda maddelerinin korunmasında kullanılan nitrozamin türevleri, içme suyundaki nitritler ve inek sütü bulunmaktadır (27).
Tip 1 diyabetin asemptomatik olduğu preklinik dönemde dolaşımda adacık hücre antikorları (ICA) saptanabilir. Humoral immün belirteçlerden en önemlileri GAD65, insülin ve ICA512 ( Islet cell antigen 512)’dir (28). Preklinik dönem, bebeklerde ve çocuklarda çok kısa iken, ileri yaşlarda aylarca hatta yıllarca sürebilir (15).
Tip 1 diyabet genellikle 30 yaş öncesinde başlar. Görülme sıklığında, okul öncesi (6 yaş civarı), puberte (13 yaş civarı) ve geç adolesan dönem (20 yaş civarı) olmak üzere üç pik görülür. Daha ileri yaşlarda ortaya çıkabilen latent otoimmün diyabet formunun, çocukluk çağı (15 yaş altı) tip 1 diyabete yakın oranlarda görüldüğü bildirilmektedir (29).
Erken klinik dönemde hiperglisemiye ilişkin noktüri, poliüri, polidipsi, yorgunluk, ağız kuruluğu gibi semptomlar görülür. Tanı ve tedavisi gecikmiş hastalarda ketoasidoz bulguları ortaya çıkabilir. Bu dönemde beta hücre rezervi azalmıştır. Açlık C-peptid düzeyleri 0.5 ng/mL’nin üzerindedir. Klinik semptomların tam olarak yerleştiği dönemde beta hücre rezervleri çok düşüktür (C-peptid < 0.1 ng/mL). Otoantikor titreleri azalır. Hastalar mutlak ekzojen insülin gereksinimi gösterirler ve glisemi ayarı güçtür. Ketoasidoz, hipoglisemi gibi akut komplikasyonlara daha sık rastlanır. İleri evrede mikrovasküler ve makrovasküler komplikasyonlar gelişir. Hastaların yaklaşık % 50’sinde ölüm nedeni diyabetik nefropatidir (26).
2.1.6. Tip 2 Diabetes Mellitus
Diyabet hastalarının % 80-90’ını tip 2 diyabet hastaları oluşturur. Hastaların çoğu obez veya kiloludur. Hastalık çoğunlukla 30 yaşından sonra ortaya çıkar, ancak obezitenin yaygınlaşması nedeniyle, son 10-15 yıldır çocuk ve adölesanlarda görülme sıklığı artmıştır. Hastalık genellikle sinsi başlangıçlıdır. Pek çok hastada başlangıçta hiçbir semptom görülmez. Bazı hastalar ise, tekrarlayan mantar enfeksiyonları, yara iyileşmesinde gecikme, bulanık
9
görme, el ve ayaklarda uyuşma, ayak ağrıları gibi kronik komplikasyonlara bağlı bulgular ile başvurabilir (29).
Tip 2 diyabet için güçlü bir genetik yatkınlık söz konusudur. Monozigot ikizlerde konkordans % 70 ile % 90 arasındadır. Her iki ebeveyni tip 2 diyabetik olan kişilerde diyabet görülme riski % 40 civarındadır. Hastalık poligenik ve multifaktöriyeldir. Genetik yatkınlığa ek olarak obezite, beslenme, fiziksel aktivite gibi çevresel faktörler fenotipi belirler. Genetik yatkınlığı belirleyen ortak bir gen gösterilememiştir. Hastalığın poligenik formlarında peroksizom proliferatör-aktive reseptör gama (PPAR-γ), intestinal yağ asidi bağlayıcı protein 2 (FABP2), Calpain 10 ve β3 adrenerjik reseptör mutasyonları tanımlanmıştır (16, 30).
Tip 2 diyabet, bozulmuş insülin sekresyonu, insülin direnci, hepatik glukoz üretiminde artış ve anormal yağ metabolizması ile karakterizedir. İnsülin direnci, insülinin hedef dokuları olan karaciğer, kas ve yağ dokusunda ekzojen veya endojen insüline karşı biyolojik yanıtın bozulmasıdır. İnsülin karaciğerde glukoneogenezi ve glikojenolizi inhibe ederek hepatik glukoz üretimini baskılar. Ayrıca, glukozun kas ve yağ dokusuna alımını ve burada enerji kaynağı olarak depolanmasını sağlar. İnsülinin bu etkilerine karşı direnç gelişimi, hepatik glukoz çıkışında artış (hepatik insülin direnci), glukozun kas ve yağ dokusu içine alınamaması (periferik insülin direnci) ve hiperglisemi ile sonuçlanır. Hastalığın erken evrelerinde hiperglisemi beta hücrelerinde insülin salınımının artışı ile kompanze edilir. Beta hücrelerinin bu kapasitesi aşıldığında ise diyabet ortaya çıkar (26).
İnsülin direncine neden olan moleküler mekanizmalar tam olarak aydınlatılamamıştır. İnsülin direnci; pre-reseptör düzeyinde (anormal beta hücre salgı ürünleri, dolaşan insülin antikorları), reseptör düzeyinde (reseptör sayısında azalma, reseptör mutasyonları) veya post-reseptör düzeyinde olabilir. İnsülin direnci gelişiminden sıklıkla tirozin kinaz aktivitesinde azalma, reseptör sinyal sisteminde bozukluklar, glukoz transportunda azalma gibi post-reseptör düzeyindeki bozukluklar sorumludur (26).
Hastalığın erken evrelerinde insülin direncine yanıt olarak artan insülin salınımı, zaman içinde azalarak ağır insülin yetmezliğine kadar ilerleyebilir. İnsülin salınım kapasitesindeki bu azalmanın nedeni belirsizdir. Diyabetin neden olduğu metabolik çevrenin adacık hücre fonksiyonlarını olumsuz yönde etkilediği düşünülmektedir. Kronik hiperglisemi (glukotoksisite) ve serbest yağ asitlerinin artışı (lipotoksisite) adacık hücre işlev bozukluğunun ilerlemesine yol açar (16).
Obezite (özellikle santral ya da visseral), tip 2 DM patogenezinde önemli bir role sahiptir. İntraabdominal adipoz dokuda lipolitik aktivite yüksektir ve insülin etkisine dirençlidir.
10
Dolaşımda serbest yağ asitleri ile esterleşmemiş serbest yağ asitleri, retinol bağlayıcı protein-4, leptin, TNF-α, resistin ve adiponektin gibi adiposit ürünleri artar. Artmış serbest yağ asitleri ve adipokinler iskelet kası ve karaciğerde insülin direnci gelişmesine, beta hücre işlevlerinde bozulmaya ve inflamatuar sitokinlerin salınımına neden olabilir. Ayrıca serbest yağ asitlerinin artışı sonucunda hepatositlerde VLDL ve trigliserid sentezi artar, dislipidemi gelişir. Karaciğerde lipid depolanması, nonalkolik yağlı karaciğer hastalığı ve karaciğer fonksiyon testlerinde bozukluklara yol açabilir (16).
Tip 2 diyabetin önlenmesinde yaşam tarzı değişikliği temel unsur olarak görülmektedir. Bozulmuş açlık glukozu veya bozulmuş glukoz toleransı olan hastalarda yaklaşık % 5-10 kilo kaybı ve günde 30 dakika süreli orta şiddette fiziksel aktiviteyi içeren yaşam tarzı değişimi en uygun yaklaşım olarak kabul edilmektedir. Bozulmuş açlık glukozu ve bozulmuş glukoz toleransı birlikteliğinde ise metformin tedavisine başlanması önerilmektedir (29).
Diyabetik hastalarda tanı konduğu andan itibaren mikro ve makrovasküler komplikasyonların varlığı araştırılmalıdır. İlk yapılması gereken laboratuvar incelemeleri; HbA1c, açlık lipid profili, serum kreatinini, tiroid fonksiyon testleri, tam idrar analizi, idrarda albümin atılımının ölçümü ve EKG’dir. İdrar albumin atılımının saptanmasında sabah ilk (veya spot) idrarda albumin/kreatinin oranı tercih edilmelidir. HbA1c ölçüm sıklığı, insülin kullanan hastalarda üç ayda bir, diğer hastalarda üç-altı ayda bir olmalıdır. Göz dibi muayenesi, lipid profili, idrar albümin atılımı ve serum kreatinin ölçümleri yılda bir kez tekrarlanmalıdır. Komplikasyonların önlenmesinde glisemik kontrolün ve kan basıncı kontrolünün sağlanması esastır (29).
2.1.7. Diabates Mellitus’un Komplikasyonları
Diabetes mellitus komplikasyonları akut ve kronik olarak snıflandırılır: Akut komplikasyonlar:
Diyabetik ketoasidoz
Hiperozmolar hiperglisemik durum Laktik asidoz
11
Kronik komplikasyonlar:
Makrovasküler komplikasyonlar: o Kardiyovasküler hastalıklar o Serebrovasküler hastalıklar o Periferik damar hastalığı Mikrovasküler komplikasyonlar:
o Nefropati
o Retinopati
o Nöropati
2.1.8. Diyabetik Nefropati
Diyabetik nefropati, tip 1 ve tip 2 diyabetli hastalarda morbidite ve mortalitenin en önemli nedenleri arasındadır. Tüm dünyada son dönem böbrek yetmezliği nedenleri arasında diyabetik nefropati ilk sırada yer almaktadır. Ayrıca diyaliz uygulanan hastaların büyük bir bölümünü diyabetik hastalar oluşturmaktadır (3, 4).
Tip 1 diyabetli hastaların %30-40’ında tanıdan ortalama 20 yıl sonra nefropati ortaya çıkmaktadır. Olguların %50’sinde, klinik nefropatiyi takiben 10 yıl içinde, % 75’den fazlasında ise 20 yıl içinde son dönem böbrek yetmezliği gelişmektedir (5). Tip 2 diyabetli hastalarda ise, tanı anında nefropati oranı % 5-10 iken, 10 yıl sonrasında bu değer % 25-40’a ulaşmaktadır. Klinik nefropati gelişen olguların %20’sinde olay 20 yıl içinde son dönem böbrek yetmezliğine ilerlemektedir (5, 6).
Diğer mikrovasküler komplikasyonlarda olduğu gibi diyabetik nefropati patogenezinde de kronik hiperglisemi temel rolü oynamaktadır. Diyabetik nefropati gelişimi için en önemlisi hipergliseminin süresi ve şiddeti olmak üzere, genetik yatkınlık, hipertansiyon, dislipidemi, sigara kullanımı, fazla protein alımı gibi risk faktörleri tanımlanmıştır (31). Nefropati başladıktan sonra tablonun ilerlemesine yol açan en önemli risk faktörü ise hipertansiyondur. Kan basıncı kontrolünde, antiproteinürik etkileri nedeniyle tercih edilen ACE inhibitörü ve anjiyotensin reseptör blokeri ajanlar, renal hasarlanmadan korunmada ve nefropatinin progresyonunu azaltmada etkilidirler. (6, 31-33).
12
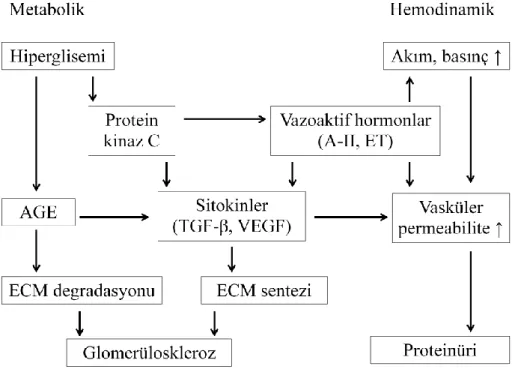
Diyabetik Nefropati Patogenezi :
Diyabetik nefropati, diyabetin erken dönemlerinde başlayan hemodinamik değişiklikler ile, hipergliseminin tetiklediği metabolik faktörlerin etkileşimi sonucunda gelişir. Çeşitli sitokinler ve büyüme faktörleri de bu sürece katkıda bulunmaktadır.
Diyabete bağlı böbrek hasarının en erken bulgusu glomerüler hipertansiyon ve hiperfiltrasyondur. Afferent arteriollerde vazodilatasyon, renal kan akımı ve intraglomerüler basıncı arttırmaktadır. Glomerüllerde meydana gelen hemodinamik ve mekanik stres, endotel fonksiyonlarında bozulma ve proteinüriye neden olurken, diğer yandan glomerüloskleroz ve fibrozis ile ilişkili sitokin ve büyüme faktörlerinin salınımını uyarmaktadır (34, 35).
Diyabetik hastalarda hem glomerüllerde hem de interstisyumda lokal renin-anjiotensin-aldosteron sistemi uyarılmıştır. Doku anjiotensin II düzeylerinin artışı; oksidatif stres ürünlerinin oluşumu, endotel hasarı, vazokonstriksiyon, tromboz, inflamasyon, vasküler yeniden yapılanma ve transforme edici büyüme faktörü beta (TGF-β) aracılı hücre dışı matriks birikimi ile sonuçlanır (36).
Diyabetik nefropati gelişiminde en önemli faktör kronik hiperglisemi ile ilişkili metabolik değişikliklerdir. Hiperglisemi, glukotoksisite, polyol yolu aktivasyonu, protein kinaz C aktivasyonu, ileri glikasyon ürünlerinin artışı gibi biyokimyasal değişiklikler glomerül geçirgenliğinde değişikliklere ve matriks proteinlerinin artışına yol açar (37).
Hiperglisemi, endotel ve epitel hücrelerinde direkt toksik etkileri ile hücre dışı matriks birikimine neden olan kollajen, fibronektin, laminin, TGF-β1 sentezini arttırır. Ayrıca mezenjial hücrelerde heparan sülfat sentezini azaltarak proteinüriye katkıda bulunur (38). Böbrekler, lens ve retina gibi, glukoz alımının insülinden bağımsız olduğu dokularda glukoz fazlası aldoz redüktaz enzimi aracılığı ile sorbitole, sorbitol de sorbitol dehidrogenaz enzimi ile fruktoza dönüştürülür. Kronik hiperglisemide polyol yolunun aktive olması hücre içi sorbitol birikimine neden olur. Artan sorbitolün etkisiyle miyoinositol ve Na+
-K+ATP’az aktivitesi azalır, ozmoregülasyon bozulur. Ayrıca NADPH tüketimi oksidatif stresi arttırır (39, 40). Deneysel diyabet modellerinde bu yolun mikrovasküler komplikasyonların gelişiminde önemli rolü olduğu ve aldoz redüktaz enzimi inhibitörleri ile önlenebildiği gösterilmiştir (41).
Serin-treonin kinaz grubu bir enzim olan protein kinaz C’nin hiperglisemiye sekonder olarak aktivasyonu, komplikasyonların gelişiminde önemli bir role sahiptir. Protein kinaz C aktivasyonu ile fosfolipaz A2 aktivite artışı ve bununla birlikte salınımı artan araşidonik asit,
13
prostaglandinler ve tromboksan A2, hiperfiltrasyon ve kan basıncı değişikliklerine verilen renal yanıtı değiştirmektedir (42, 43). Protein kinaz C aktivasyonu, TGF-β1 sentezini uyararak mezenjial hücrelerde ekstrasellüler matriks yapmını arttırır. Deneysel çalışmalarda protein kinaz C artışının, vasküler endotelyal büyüme faktörü (VEGF) aracılığıyla glomerüler permeabilite artışına ve mezenjial hücre aktivasyonu ile glomerüloskleroza neden olduğu gösterilmiştir (44, 45).
Mikrovasküler komplikasyonların patogenezinde rol oynayan diğer bir faktör, proteinlerin nonenzimatik glikozillenmesi ve ileri glikasyon son ürünlerinin (AGE) birikimidir. Yüksek glukoz düzeylerinde, glukoz non-enzimatik olarak amino guruplarına bağlanır ve Schiff bazları oluşur, daha sonra hala reversibl olan Amadori ürünleri ve Amadori bileşiklerinin çapraz bağlar oluşturmasıyla stabil AGE’ler oluşur (46). AGE’lerin bağlandığı spesifik reseptörler (RAGE) retina, periferik sinirler, vasküler endotelyal hücreler, renal proksimal tübüler hücreler ve mezenjial hücrelerde bulunur (47). AGE’ler diyabet komplikasyonlarında temelde iki şekilde rol almaktadır. Birincisi; özellikle ekstrasellüler matriksin yapısındaki proteinler arasında çapraz bağlar oluşturarak matriks yapısını ve fonksiyonlarını bozması, ikincisi ise; kendine özgü reseptörlerine bağlanması sonucunda çeşitli sinyal yolaklarını aktive ederek çok sayıda sitokin, kemokin ve vazoaktif hormon üretimini uyarmasıdır. AGE’ler, böbreklerde mezenjial matriks artışına ve TGF-β salınımını uyararak bazal membran kalınlaşmasına neden olur (48).
Erken dönemde glomerülopati oluşumunda etkili büyüme faktörleri; vasküler endotelyal büyüme faktörü (VEGF), transforme edici büyüme faktörü beta (TGF-β), büyüme hormonu (GH), insülin benzeri büyüme faktörü-1 (IGF-1) ve bağ dokusu büyüme faktörü (CTGF)’dür (49). Özellikle VEGF ve TGF-β, patogenezde merkezi bir role sahiptir. Böbrekte VEGF podositlerden eksprese edilir. VEGF reseptörleri özellikle glomerüler endotel hücreleri üzerinde bulunur ve endotel hücrelerinden anti-apoptotik proteinlerin salınımına yol açar. Hipoksi, hiperglisemi, anjiotensin II, protein kinaz C, AGE gibi uyaranlarla VEGF salınımının artması glomerüler hipertrofi ile sonuçlanır (50, 51). Diyabetik hastalarda glomerüler TGF-β ekspresyonu, kötü glisemik kontrol ile orantılı olarak yüksek bulunmuştur (52). TGF-β ekspresyonu erken evrelerde hiperglisemi ve protein kinaz C, ileri evrelerde ise glikolize proteinler ve oto-indüksiyon ile uyarılır. TGF-β tip 1 kollajen, tip 4 kollajen, fibronektin, laminin gibi ekstrasellüler matriks moleküllerinin yapımını arttırarak glomerüler hipertrofi ve mezenjial matriks birikimine neden olmaktadır (53, 54).
14
Diyabetik hastalarda renal hasarlanmanın nedenlerinden biri de oksidatif strestir. Yüksek glukoz düzeyleri glomerüler mezenjial ve tübüler epitelyal hücrelerde reaktif oksijen türleri (ROS) oluşumunu arttırmaktadır. Yüksek glukoz düzeylerinde ROS’un en önemli kaynağı mitokondriyal metabolizma ve NADPH oksidaz’dır. Ayrıca AGE, sitokinler ve protein kinaz C de NADPH oksidaz ve ROS oluşumunu uyarmaktadır. ROS, sinyal transdüksiyon kaskadı ve transkripsiyon faktörlerini aktive ederek TGF-β ve ekstrasellüler matriks proteinlerinin yapımını arttırır (54). ROS artışı ile birlikte hücre zarı lipid peroksidasyonu, protein oksidasyonu ve DNA hasarı meydana gelir. Lipid peroksidasyonu ile oluşan prostaglandinler, renal vazokonstriksiyona ve nefronda hemodinamik değişikliklere yol açmaktadır (55, 56).
Şekil 2.1. Diyabetik nefropati patogenezi. A-II: anjiotensin-II, ET: endotelin, AGE: ileri glikasyon ürünleri, TGF-β: transforme edici büyüme faktörü beta, VEGF: vasküler endotelyal büyüme faktörü, ECM: ekstrasellüler matriks (57).
Diyabetik Nefropatide Morfolojik Değişiklikler:
Diyabetik nefropatide tüm renal kompartmanlarda yapısal değişiklikler meydana gelmektedir. Karakterisik glomerüler lezyonlar kapiller bazal membran kalınlaşması, diffüz glomerüloskleroz ve nodüler glomerülosklerozdur (46). Diyabet gelişiminin ilk yıllarında glomerüler hiperperfüzyon ve renal hipertrofi, beş yıl içinde glomerüler bazal membran (GBM) kalınlaşması ve mezenjial genişleme meydana gelir. GBM kalınlaşması erken dönemde ancak elektron mikroskobu ile saptanabilirken, normalin 2-3 katına ulaştığında ışık
15
mikroskobu ile görülebilir. Zamanla mezenjial genişleme glomerüler kapillerleri sınırlar, yapılarında bozulmaya neden olur (58).
Mezenjial hücre proliferasyonu ve mezenjial matrikste diffüz artış diffüz glomerüloskleroz olarak tanımlanır ve her zaman bazal membran kalınlaşması ile ilişkilidir. En sık görülen histolojik bulgu diffüz interkapiller glomerülosklerozdur. Hastaların % 40-50’sinde periferik glomerüler lobüllerin merkezinde büyük asellüler nodüller oluşur (Kimmelstiel-Wilson lezyonu). Bu lezyonlar renal hastalığın şiddeti ile iyi korelasyon göstermemekle birlikte, diyabet için patognomonik olarak kabul edilirler (46, 57).
Diyabetik nefropatide sıklıkla hızlanmış renal arteriyoskleroz ve arteriyoloskleroz görülür. Büyük arterlerde meydana gelen ateromatöz değişiklikler iskemik parankimal atrofiye sebep olarak böbrek yetmezliği gelişimine katkıda bulunabilmektedir. Renal arteriyol duvarlarında hiyalen madde birikimi belirgindir. Hiyalen arteriyoskleroz, afferent ve efferent arteriyolleri tutar ve hipertansiyon gelişiminde rol oynadığı düşünülmektedir. Efferent arteriyoskleroz diyabeti olmayan kişilerde nadir görülen bir lezyondur (42, 46, 57-59).
Tübüller, glomerülün efferent arteriyolleri tarafndan beslenmektedir. Glomerülosklerozda, tübüler iskemi, interstisyel fibrozis ve tübüler bazal membranda kalınlaşma sık görülen değişikliklerdir. Diyabetik hastalarda akut ve kronik pyelonefrite yatkınlık artmıştır. Ateş, şok, yan ağrısı, belirgin hematüri, piyüri, oligüri ve böbrek yetmezliği ile seyreden papiller nekroz, diyabetik hastalarda normal popülasyona göre çok daha sık görülmektedir. Bunun nedeni vasküler değişikliklere bağlı ortaya çıkan iskemi ve bakteriyel enfeksiyona yatkınlıktır. Ayrıca papiller nekroz, fenasetin kullanımı ile ilişkili bulunduğundan diyabetik hastalarda bu ilacın kronik kullanımdan kaçınılmalıdır (57, 58).
Diyabetik Nefropatinin Klinik Seyri:
Diyabetik nefropatinin gelişim basamakları, gelişme süreleri farklı olmakla beraber tip 1 ve tip 2 diyabetli hastalarda benzerlik göstermektedir. Diyabet gelişiminden itibaren glomerüler filtrasyon hızı ve serum kreatinin değerlerinin değişimi şekil 2.2’de gösterilmiştir (16, 60). Erken dönemde glomerüllere gelen kan akımının artması sonucunda glomerüler hiperfiltrasyon ve hipertrofi meydana gelir. Böbrek ağırlığı ve boyutları artar. GFR’de % 20-50 oranında artış görülür. Glisemik kontrolün kısa sürede (1-2 hafta) sağlanması ile böbrek boyutlarında küçülme olmaksızın, GFR normale dönebilir. Tip 1 diyabetli hastalarda yoğun insülin tedavisinin başlamasından sonra böbrek boyutlarında anlamlı bir azalma izlenebilir.
16
Aşikar diyabetin ortaya çıkışını takiben, uzun süreli ve böbrek işlev bozukluğuna ait herhangi bir laboratuvar bulgusunun bulunmadığı sessiz bir periyot başlar. Bu dönemde böbrek biyopsisi yapılacak olursa, glomerüler bazal membranda kalınlaşma ve mezengial matriks artışı görülebilir. Böbrekte yapısal değişiklikler geliştiği halde, henüz mikroalbüminüri bulunmaz (57).
Şekil 2.2. Diyabetik nefropatinin doğal seyri (16, 60).
Nefropati varlığınının en erken kanıtı mikroalbuminüridir. Albumin atılım hızı; 24 saatlik idrarda, 4 veya 8 saatlik idrarda veya spot idrarda albumin/kreatinin oranının ölçümü ile saptanabilir. Uygulama kolaylığı açısından spot idrarda albumin/kreatinin oranının ölçümü tercih edilmektedir. Albumin atılımının diürnal varyasyon göstermesi nedeniyle en uygun örnek sabah ilk idrarıdır. Albumin atılım hızlarına ait tanımlamalar tablo 2.4’te gösterilmiştir (61).
Tablo 2.4. Albumin atılım hızlarının tanımı (61).
Spot idrar (mg/g kreatinin) 24 saatlik idrar (mg/24 saat) Süreli ölçüm (µg/dakika) Normal <30 <30 <20 Mikroalbuminüri 30-299 30-299 20-199 Makroalbuminüri ≥300 ≥300 ≥200
Tip 1 diyabette, mikroalbuminüri varlığı aşikar proteinüri (≥ 300 mg/gün) gelişimi için önemli bir risk faktörüdür. Müdahale edilmemesi durumunda mikroalbuminürisi bulunan tip 1 diyabetli hastaların % 80’i, idrar albümin atılımında yıllık % 10-20 artış ile birlikte 10-15 yıl içinde aşikar nefropatiye ilerler. Mikroalbüminürinin ve kan basıncında yükselmenin
17
başladığı evre, henüz filtrasyon kaybı olmaması ve profilaktik tedaviye yanıtın iyi olması nedeniyle özel bir önem taşımaktadır. Bu dönemde dipstik testlerle pozitif sonuç elde edilemez, bu nedenle, mikroalbumin tayini duyarlı yöntemler ile yapılmalıdır.
Tip 1 diyabetik olgularda nefropati genellikle 5-10 yılda başlarken, tip 2 diyabetik olgularda, tanı anında mikroalbuminüri veya makroalbuminüri bulunabilir. Tip 2 diyabette mikroalbuminüri varlığı, makroalbuminüri için tip 1 diyabette olduğu kadar güçlü bir belirleyici olmasa da müdahale edilmezse 20 yıl içinde %20-40’ı aşikar nefropatiye ilerler. Ayrıca tip 2 diyabette albüminüri, hipertansiyon, konjestif kalp yetmezliği, prostat hastalıkları gibi diyabet dışı nedenlerle de ilişkili olabilir (16, 26, 61, 62).
Aşikar nefropati döneminde, kapiller lümen genişlemiş mezenjial matriks tarafından sınırlanmış, glomerüllerde skleroz başlamıştır. GFR, kişiden kişiye değişmekle birlikte yılda 5-10 ml/dk azalır. Hastaların çoğu bu evrede hipertansiftir ve hipertansiyon varlığı prognozu kötüleştirir. Serum kreatinini normal veya hafif artmıştır. Aşikar nefropati sıklıkla retinopati, nöropati, ve periferal ve koroner vaskülopati ile birliktelik gösterir (63-65).
Aşikar nefropatisi olan tip 1 diyabetli hastaların %50’sinde 10 yıl içinde, %75’ten fazlasında ise 20 yıl içinde SDBY gelişmektedir. Tip 2 diyabetli hastalar için bu oran 20 yıl sonrasında %20 civarındadır (61). Tedavi edilmeyen proteinüri ortalama 7 yıl içinde SDBY’ne ilerlemektedir. Bu evrede herhangi bir nedenle oluşan SDBY ile benzer şekilde, GFR ileri derecede azalmış, böbrek yetmezliğine ait semptomlar belirgin hale gelmiştir (64, 65).
Albuminüri, SDBY yanı sıra hem tip 1 hem tip 2 diyabetik hastalarda kardiyovasküler morbidite ve mortalite açısından da yüksek risk oluşturur. ADA, tip 1 diyabette tanı konulduktan 5 yıl sonra, tip 2 diyabette ise tanı anında mikroalbuminüri taraması yapılmasını ve sonrasında testin yılda 1 kez tekrarlanmasını önermektedir. Mikroalbuminüri saptanması halinde idrar yolu enfeksiyonu, hematüri, ateşli hastalıklar, şiddetli hiperglisemi, şiddetli hipertansiyon, aşırı egzersiz gibi albumin ekskresyonunda geçici artışa neden olan durumlar dışlanmalı ve test 3-6 ay içinde 2 kez tekrar edilmelidir. 2 veya 3 ölçümde artmış değerler mikroalbuminüri olarak kabul edilir (66, 67).
Diyabetik nefropatinin kesin tanısı renal biyopsi ile konulmaktadır. Ancak rutin takiplerde biyopsi uygulanması önerilmemektedir. Makroalbuminüri ile birlikte diyabetik retinopatinin bulunması tanıyı güçlendirmektedir (68). Diyabetik nefropatili hastalarda nefritik idrar sedimenti saptanması, diyabet dışında böbrek hastalığı öyküsü, proteinüride hızlı artış, günde 5 gramın üzerinde proteinüri varlığı, retinopati yokluğunda albuminüri, açıklanamayan hızlı
18
böbrek fonksiyon kaybı ve proteinüri yokluğunda renal fonksiyonlarda azalma böbrek biyopsisi endikasyonu oluşturan durumlardır (26).
Tablo 2.5. Diyabetik nefropati evreleri (63).
I. Hiperfiltrasyon evresi (tanı anında) Böbrek ve glomerüller büyüktür.
GFR %20-50 oranında artmıştır. Belirgin albuminüri yoktur. Kan basıncı normaldir.
II. Latent evre (1.5-5 yıl sonra) Glomerüler bazal membran kalındır.
GFR normaldir veya %20-50 oranında artmıştır. 15-20 μg/dk albuminüri görülür.
Kan basıncı normaldir, yılda 1 mmHg artmaya başlar. III. Yeni başlayan nefropati (5-15 yıl sonra)
Glomerüler bazal membran kalın, mezengium geniştir. GFR normaldir veya bazen cok hafif yüksektir.
20-200 μg/dk veya 30-300 mg/gün mikroalbuminüri görülür. Kan basıncı artmaya başlar, tedavi edilmezse yılda 3 mmHg artar. IV. Aşikar nefropati (10-20 yıl sonra)
Diffüz interkapiller glomerüloskleroz ve mezengiyal genişleme görülür. GFR azalmıştır (yılda 5-10 mL/dk azalır).
İlerleyici proteinüri görülür (>500 mg/gün). Kan basıncı artmıştır (yılda 5 mmHg artar).
V. Son dönem böbrek yetmezliği (> 20 yıl sonra) Belirgin glomerüloskleroz vardır.
GFR 10 ml/dk’dan azdır.
Glomerüloskleroz gelişince proteinüri azalabilir. Kan basıncı çok yüksektir.
19
2.2. KALSİYUM - FOSFOR METABOLİZMASI
2.2.1. Kalsiyum
Kalsiyum, vücutta en çok bulunan beşinci elementtir. Sağlıklı bir erişkinde, toplam 1.0-1.3 kg kalsiyum bulunmaktadır. Bu miktarın % 99’u hidroksiapatit formunda kemik dokuda, %1’i ise hücre dışı sıvı ve yumuşak dokuda bulunmaktadır. Kemik dokudaki kalsiyumun %1 kadarı serberst olarak hücre dışı sıvıya geçebilmektedir. Kemik dokuda kalsiyum birikimi fetal hayatın 3. trimestırında belirgin hale gelmekte, çocukluk ve ergenlikte hız kazanarak, erken erişkinlikte en üst düzeye ulaşmakta, sonrasında ise giderek azalmaktadır (69, 70).
Kalsiyum, kemik mineralizasyonu yanı sıra pıhtılaşma, nöral iletim, enzim aktivitesi, hormon salınımı, kas kasılması ve plazma membran potansiyelinin devamlılığı gibi birçok temel fizyolojik süreçte önemli role sahiptir. Hücresel işlevlerdeki kritik önemi nedeniyle, hücre dışı sıvıda iyonize kalsiyum konsantrasyonu dar bir aralıkta tutulmaktadır. Hücre içi serbest kalsiyum seviyesi yaklaşık 100 nmol/L’dir ve hücre dışı serbest kalsiyum konsantrasyonundan 10.000 kat daha düşüktür (71). Bu gradyent enerji bağımlı sodyum-kalsiyum pompası ile korunmaktadır. Hücre içindeki serbest sodyum-kalsiyum, ikincil mesajcı olarak hücre büyümesi, transkripsiyon, hücre bölünmesi, hücresel aktivite, sentezlenen ürünlerin salgılanması gibi süreçlerde sinyal iletimini sağlar. Hücre dışı serbest kalsiyum, total vücut kalsiyumunun küçük bir kısmını oluşturmasına rağmen, nöromüsküler ve kardiyak uyarı üzerinde önemli etkilere sahiptir. Düz kaslar, iskelet ve kalp kası, nöronlar, endokrin bezler, gastrik mukoza, lökositler ve trombositlerde bulunan voltaj kapılı kalsiyum kanalları plazma membranının depolarizasyonuna cevap olarak açılmakta ve kalsiyum hızla sitozole salınmaktadır (72, 73).
Kanda kalsiyumun yaklaşık %50’si fizyolojik olarak aktif form olan serbest veya iyonize formda, %40’ı proteine bağlı, %10’u ise bikarbonat, laktat, fosfat, sitrat gibi çeşitli anyonlar ile kompleks halindedir. Proteine bağlı kalsiyumun %80’i ise albumine bağlıdır. Serum protein konsantrasyonundaki değişiklikler, iyonize kalsiyum konsantrasyonu normal aralıkta kalsa bile, total kalsiyum düzeylerini etkiler.
Kalsiyum protein üzerindeki negatif yüklü bölgelere bağlandığından, bağlanma ortam pH’sına bağlıdır. Hücre dışı sıvıda pH değişimi sonucunda, total serum kalsiyumu değişmeden üç formun dağılımı değişebilir. Alkalozda proteinlere bağlanmanın artması nedeniyle serbest
20
kalsiyum seviyeleri azalır, asidozda ise proteinlere bağlanma azalır ve serbest kalsiyum seviyeleri artar (70, 71).
Yetişkinlerde total kalsiyum düzeyleri 8.6-10.3 mg/dL, iyonize kalsiyum ise 4.64-5.28 mg/dL arasındadır. Serbest kalsiyum ölçümü klinik olarak daha yararlı olmasına karşın, henüz total kalsiyum ölçümünün yerini almamıştır. Total kalsiyum ölçümü, proteine bağlı ve iyonize kalsiyum ölçümünü içermektedir. Hipoalbuminemi serum kalsiyum sonuçlarının hatalı düşük bulunmasına neden olabilmektedir. Serum albumininde her 1 gr/dL azalma için total serum konsantrasyonuna 0,2 mmol/L ya da 0,8 mg/dL eklenerek düzeltilmiş kalsiyum seviyesi hesaplanmalıdır. [Düzeltilmiş kalsiyum (mg/dL) = Total kalsiyum + 0.8 (4-albumin)]. Hesaplama yerine iyon selektif elektrod ile serbest kalsiyum miktarının ölçülmesi önerilmektedir (70, 71).
Diyet ile alınan günlük kalsiyum miktarı yaklaşık 600-800 mg’dır. Osteoporozdan korunmak için erişkinlerin 1200 mg/gün, laktasyon dönemi ve östrojen tedavisi almayan postmenopozal kadınların ise 1500 mg/gün elemental kalsiyum alması önerilmektedir (74).
Yetişkinlerde diyet ile alınan kalsiyumun yarısından daha azı emilmektedir. Emilim büyüme döneminde, gebelikte ve laktasyonda artmakta, ilerleyen yaşlarda azalmaktadır. İntestinal kalsiyum emiliminde aktif (transsellüler) ve pasif (parasellüler) mekanizmalar rol oynamaktadır. Parasellüler kanallardan difüzyonla kalsiyum geçişi günlük kalsiyum alımının %5’ini oluştururken, esas olarak 1,25(OH)2D tarafından kontrol edilen aktif mekanizma
alımın %20-70’inden sorumludur (71, 75). Aktif kalsiyum transportu duodenum ve proksimal jejunumda TRPV1-6 (transient reseptör potansiyelli-vanilloid iyon kanalı), Na+-Ca2+ pompası ve Ca2+-Mg2+ bağımlı ATPaz kalsiyum kanalı aracılğı ile gerçekleşmektedir. Kalsiyum iyonları epitel hücrelerinin apikal membranlarında bulunan kalsiyum kanalları (TRPV5 ve TRPV6) aracılığıyla epitel hücrelerine giriş yapmakta ve hücre içinde vitamin D’ye bağımlı bir protein olan kalbindin D’ye bağlanmaktadır. Kalbindin D proteinine bağlı olarak apikal membrandan bazolateral membrana taşınan kalsiyum iyonları, Na+-Ca2+ pompası ve Ca-ATPaz enzimi yardımıyla epitel hücrelerinden dolaşıma aktarılmaktadır. Renal tubüler epitel hücrelerinde de kalsiyum transportu benzer şekilde gerçekleşmektedir (Şekil 2.3), (76).
İntestinal kalsiyum emilimi için temel uyaran D vitaminidir. 1,25(OH)2D bu etkisini vitamin
D reseptörüne (VDR) bağlanıp TRPV6 ve lüminal kalsiyum kanallarının ekspresyonunu arttırarak gerçekleştirir. PTH, böbreklerde 1α-hidroksilaz aktivitesini ve buna bağlı olarak 1,25(OH)2D sentezini uyararak, indirekt olarak intestinal kalsiyum emilimini arttırır. Ayrıca
21
arttırır. İntestinal içerikte kalsiyum fosfor oranının 2:1’den büyük olması çözünmeyen kalsiyum fosfat oluşumuna yol açarak kalsiyum emilimini inhibe eder. Diyet ile alınan fitik asit, okzalat ve yağ asidleri de çözünmeyen kalsiyum bileşikleri oluşturabilir. Kalsiyum emilimini inhibe eden diğer faktörler intestinal içeriğin aşırı derecede alkali olması, glukokortikoidler ve tiroid hormonlarıdır (72, 75).
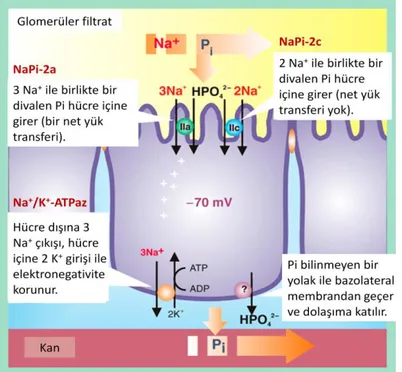
Şekil 2.3. İntestinal ve renal tubüler epitel hücrelerinde kalsiyum transportu (76).
Glomerüllerden günde yaklaşık 8-10 gram kalsiyum filtre edilir, ancak bunun yalnızca %2-3 kadarı idrar ile atılır. Diyet ile kalsiyum alımındaki değişimlerin idrar yolu ile kalsiyum atılımına etkisi azdır. Glomerüllerden filtre edilen kalsiyumun büyük bir bölümü (%65) proksimal tubüllerde sodyum-potasyum-klor (Na+
-K+-2Cl-) taşıyıcısı ile parasellüler olarak geri emilir. Bu geri emilimde, spesifik düzenleyici bir mekanizma bulunmamaktadır. Glomerüler filtrattaki kalsiyumun %20’si Henle kulpunun çıkan kalın kolunda yine parasellüler yol ile geri emilir, ancak bu segmentteki geri emilime parasellin-1 adı verilen bir
tight-junction proteini aracılık eder. Kanda kalsiyum veya magnezyum düzeyleri
yükseldiğinde, Henle kulpunun çıkan kalın kolunda kalsiyum duyarlı reseptörler (CaSR) aktive olur, Na+-K+-2Cl- taşıyıcısı inhibe edilir ve lümen pozitif voltajı azaltılarak kalsiyumun renal geri emilimi azaltılır. CaSR’nin bu işlevi sonucunda, serum iyonize kalsiyum düzeyi, PTH ve 1,25(OH)2D’den bağımsız olarak kalsiyum geri emilimini düzenler. Filtre edilen
kalsiyumun %10’u ise, transsellüler bir mekanizma ile distal tubüllerde geri emilir. Transsellüler geri emilim intestinal epitel hücrelerinde olduğu gibi, 1) kalsiyumun apikal yüzeyden TRPV5 ile hücreye girişi, 2) kalbindin D’ye bağlanarak bazolateral membrana
22
taşınması, 3) Na+-Ca2+ pompası ve Ca-ATPaz enzimi aracılığı ile dolaşıma aktarılması şeklinde gerçekleşir (Şekil 2.3). Bu işlem PTH, PTHrP ve 1,25(OH)2D tarafından uyarılır
(71).
Ekstrasellüler sıvı hacminin azalması, hipokalsemi, metabolik alkaloz ve tiazid grubu diüretiklerinin kullanımı renal tubüler kalsiyum geri emilimi arttırırken; ekstrasellüler sıvı hacminin artması, hiperkalsemi, hiperfosfatemi, metabolik asidoz ve loop diüretiklerinin kullanımı idrarla kalsiyum atılımını arttırmaktadır. Büyüme hormonu, tiroid hormonu, glukokortikoidler, mineralokortikoidler ve glukagon ise, idrar yolu ile kalsiyum atılımını arttıran hormonal faktörlerdir (77).
Ekstrasellüler sıvıda iyonize kalsiyum düzeyi, PTH ve 1,25(OH)2D tarafından
düzenlenmektedir (Şekil 2.4). Bu hormonların hedef organları kemik, böbrek ve bağırsaktır. Paratiroid bezi şef hücrelerinde bulunan CaSR’ler serum iyonize kalsiyum düzeyinde meydana gelen en ufak değişiklikleri algılamakta ve kısa sürede PTH salgılanmasını düzenlemektedir. Plazmada iyonize kalsiyum düzeyi azaldığında, paratiroid hücrelerinde CaSR inaktive olmakta ve PTH sekresyonu artmaktadır. PTH, osteoblast ve prekürsörlerinde tümör nekroz faktör süper ailesinin bir üyesi olan RANKL (reseptör aktivatör nükleer kappa B ligand) üretimini uyarır. RANK (reseptör aktivatör nükleer kappa B) aktivasyonu sonucunda osteoklast oluşumu ve aktivitesi artar, apoptoz ise baskılanır. Kemik matriksindeki rezorbsiyon, ekstrasellüler sıvıya kalsiyum ve fosfat salınımına yol açar. PTH aynı zamanda distal nefronda üriner fosfat atılımı ve kalsiyum geri emilimini uyarır. Bu mekanizmaların düzenli olarak işleyebilmesi için yeterli miktarda 1,25(OH)2D faaliyeti gereklidir. Kemik
rezorbsiyonu ve üriner kalsiyum geri emiliminin artışı ile serum kalsiyum seviyesini dengelenmesi, dakikalar veya saatler içerisinde meydana gelen bir PTH yanıtıdır. Plazma PTH düzeyi uzun süre yüksek kalırsa, böbrekte 1α-hidroksilaz ekspresyonunun artması ile 1,25(OH)2D sentezi ve salınımı artar. 1,25(OH)2D gastrointestinal sistemden kalsiyum ve
fosfat emilimini arttırır. Buna karşın serum iyonize kalsiyum konsantrasyonunda meydana gelen artış paratiroid bezinde CaSR’yi aktive eder. Bu aktivasyonun sonucunda PTH sentezi ve salınımı inhibisyona uğrar. PTH düzeyinde meydana gelen düşmeye bağlı olarak idrarda kalsiyum atılımı artar, kalsiyum havuzundan daha az kalsiyum salınımı olur. Eş zamanlı olarak aktive olan distal renal tübüllerdeki CaSR bağlı olarak glomerüler filtrattaki kalsiyumun geri emilimi azalarak üriner kalsiyum atılımı artar. Böylece serum iyonize kalsiyum konsantrasyonu düşürülür. Eğer plazma PTH düzeyi uzun süre düşük kalırsa renal 1,25(OH)2D üretimi azalır, buna bağlı olarak intestinal kalsiyum emilimi de azalır. Kalsitonin
23
ise PTH ve 1,25(OH)2D etkilerine karşı düzenleyici bir hormondur; osteoklastik yıkımı inhibe
ederek serum kalsiyum ve fosfat düzeylerini azaltır. Kalsitoninin kemik metabolizması üzerindeki etkileri PTH ve vitamin D’ye göre daha zayıftır. Sekresyonları primer olarak serum kalsiyum ve fosfat düzeylerinden etkilenmeyen, ancak kalsiyum metabolizmasını etkileyen diğer hormonlar tiroid hormonu, büyüme hormonu, adrenal glukokortikoidler ve gonadal steroidlerdir (75-78).
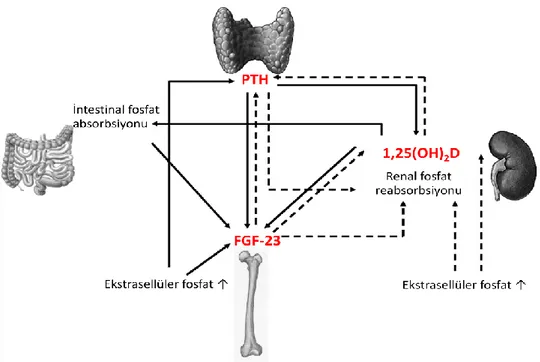
Şekil 2.4. Kalsiyum ve fosfat dengesinin PTH ve 1,25(OH)2D ile düzenlenmesi (79).
2.2.2. Fosfor
İnsan vücudunda yaklaşık 700-800 g fosfor bulunur. Bu miktarın % 80-85 kadarı, hidroksiapatit ve kalsiyum fosfat formunda kemik dokuda bulunmaktadır. Kalan %15 ise, inorganik fosfat formunda hücre dışı sıvıda ve hücre içi organik fosfat bileşikleri olarak yumuşak dokuda yer almaktadır.