T.C.
EGE ÜNİVERSİTESİ TIP FAKÜLTESİ
ÇOCUK SAĞLIĞI VE HASTALIKLARI ANA BİLİM DALI PROF. DR. KAAN KAVAKLI
KÖK HÜCRE TRANSPLANTASYONU YAPILAN TALASEMİ MAJÖR TANILI HASTALARDA
UZUN DÖNEM YAŞAM KALİTESİNİN DEĞERLENDİRİLMESİ
UZMANLIK TEZİ
Dr. OruçAli Guliyev
DANIŞMAN Prof. Dr. Serap Aksoylar
II
ÖNSÖZ VE TEŞEKKÜR
Eğitimime değerli katkılarından dolayı, birlikte çalışmaktan onur duyduğum ve akademik deneyimlerini tezimin her aşamasında benimle paylaşan saygıdeğer tez danışmanım Prof. Dr. Serap AKSOYLAR başta olmak üzere tüm Ege Üniversitesi Çocuk Sağlığı ve Hastalıkları AD Öğretim Üyeleri’ne
Tezimin oluşturulması ve hazırlanmasındaki katkı ve yardımları için Sayın Uzm. Dr. Gülcihan Özek‘e, Tez çalışmam süresince her zaman yanımda olarak bana yardımcı olan başta Sayın Prof. Dr. Savaş Kansoy olmak üzere Çocuk Kök Hücre Nakil Ünitesi ekibi’ne,
Pediatri mesleğinin inceliklerini ve deneyimlerini her fırsatta paylaşan, gerçek birer “abi” ve “abla” olan uzmanlarıma,
Tezim boyunca güleryüzleri ile yardımlarını esirgemeyen Kök Hücresi Nakil Ünitesi hemşiresi Nermin Zurnacı’ya, sekreter Nurçin Dinç’e ve biyoloğu Ayben Akcadar’a ve personelimiz Naime ablaya,
Ege Üniversitesi Tıp Fakültesi Biyoistatistik ve Tıbbi Bilişim AD öğretim üyelerine ve Arş. Gör. Gülden Hakverdi’ye,
Asistanlık süreci boyunca birlikte çalıştığım, ailemden çok vakit geçirdiğim ve çok sevdiğim tüm araştırma görevlisi arkadaşlarıma, özellikle tezimi yazarken bana hep yardım eden, asistanlığımda her anımda yanımda olan, dostlarım, canım eş kıdemlilerim Ayşe Gadaşova’ya, Lale Şeyhova’ya, Baki Beyter’e, Sinem Özdemir’e,
Asistanlık sürecini daha güzel kılan, güzel anıları paylaştığım, her zorlukta yanımda olan, bana yardımlarını esirgemeyen canım arkadaşım Mustafa Taşçı’ya,
Doğduğum günden beri ellerimden tutan, beni destekleyen, bana hayat veren, yol gösteren, bıkmadan usanmadan beni dinleyen, en yakın dostum, canım annem Gülnaz Hüseynova’ya,
Her zaman beni destekleyen ve yanımda olan sevgili ablam Türkane’ye ve sevgili kardeşim Cemil’e
Minnet ve teşekkürlerimi sunarım.
Dr. Oruç Ali GULIYEV 2019, İzmir
III İÇİNDEKİLER TABLOLAR DİZİNİ ... V KISALTMALAR ... VII ÖZET ... IX SUMMARY ... XIV 1.GİRİŞ ve AMAÇ ... 1 2.GENEL BİLGİLER ... 2 2.1.Talasemi ... 2 2.1.1. Tanım ... 2 2.1.2. Talasemide sınıflama ... 2
2.1.3. Talasemide tedavi prensipleri ... 4
2.2.Hematopoetik Kök Hücre Nakli (HKHN) ... 6
2.2.1.Tanım ... 6
2.2.2.Kök hücre kaynakları ... 8
2.2.3. Hazırlanma rejimi ... 9
2.2.4. Engraftman ... 10
2.2.5. Kök hücre nakli sonrası gelişen komplikasyonlar ... 10
2.3. Talassemide HKHN ... 21
2.4. HKHN yapılan olgularda yaşam kalitesi ... 24
3. MATERYAL METOD ... 26 4. BULGULAR ... 30 5.TARTIŞMA ... 44 6. SONUÇLAR ... 49 7. KAYNAKLAR ... 52 EKLER Ek 1. Etik Kurul Onay Belgesi ... 57
IV
EK 2. Yaş gruplarına göre yaşam kalitesi puanları ... 59
Ek 3. Olgu rapor formları ... 62
Ek 4. Benzeşim Onay Formu ... 88
V
TABLOLAR DİZİNİ
Tablo 1. Talassemik değerlendirme ... 7
Tablo 2. Yaygın Kullanılan Ajanlara ve Kombinasyonlara Göre Miyeloablatif ve Miyeloablatif Rejim Örneği ... 9
Tablo 3. Akut GVHH’nın klinik belirtileri ... 12
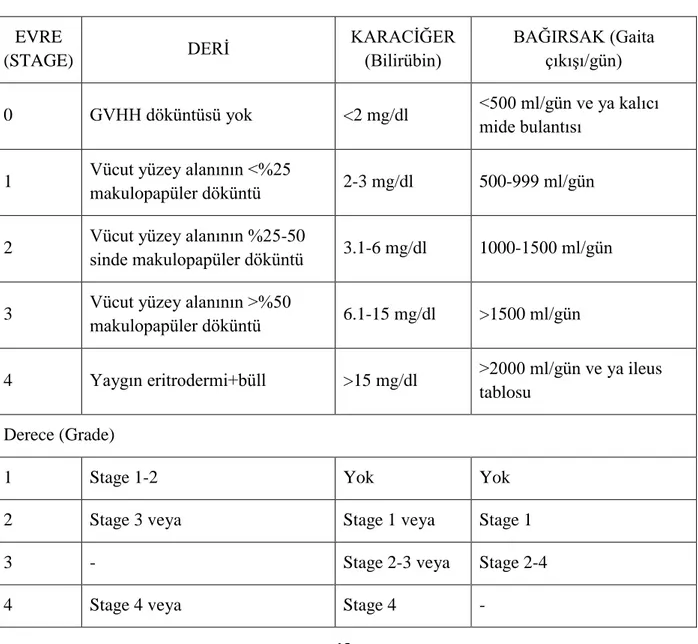
Tablo 4. Akut GVHH’nda evrelendirme... 12
Tablo 5. Graft yetmezliği tipleri ... 15
Tablo 6. KGVHH de klinik derecelendirme skorlaması ... 17
Tablo 7. Kronik GVHH’nın semptom ve belirtileri ... 18
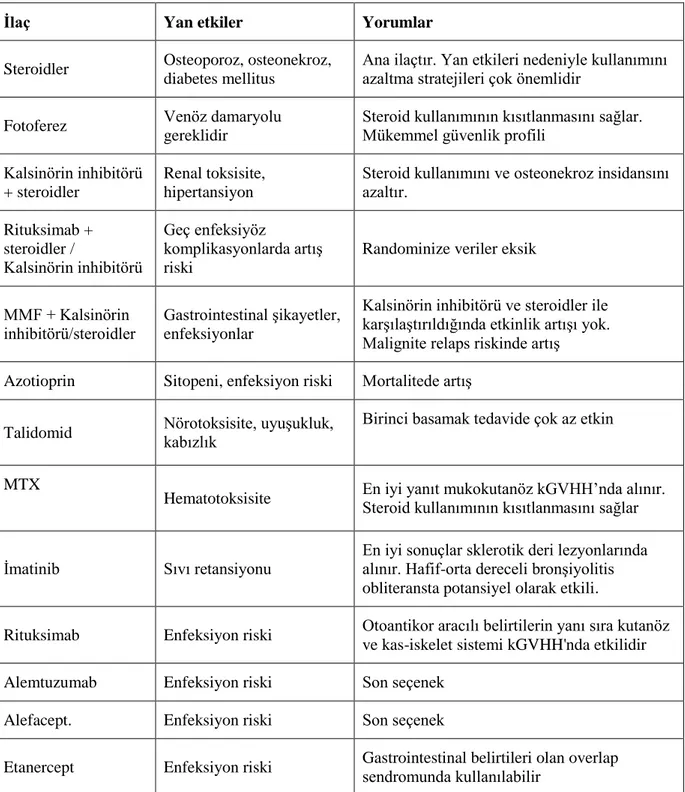
Tablo 8. Kronik GVHH’nda tedavi seçenekleri... 19
Tablo 9. Talasemili olgularda HKHN sonrası oluşan geç dönem komplikasyonların özeti... 23
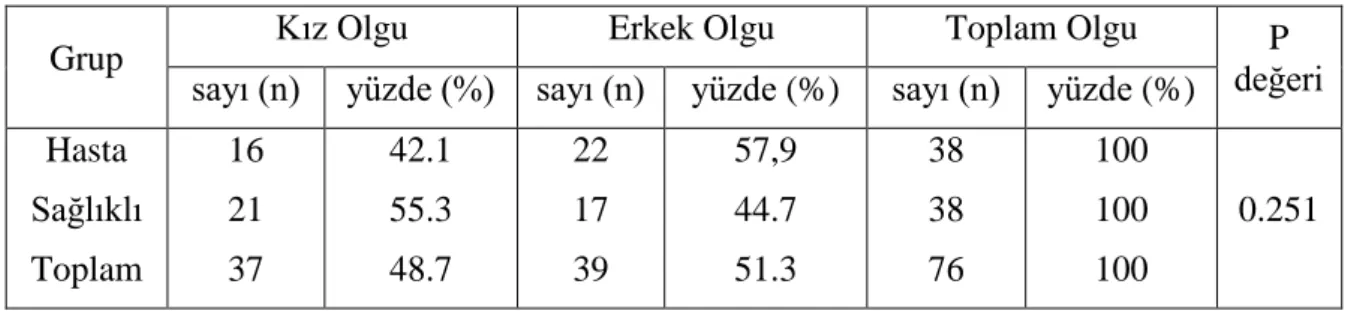
Tablo 10. Çalışmaya katılan olguların cinsiyet dağılımı ... 30
Tablo 11. Çalışmada yer alan olguların yaş gruplarına göre dağılımı ... 30
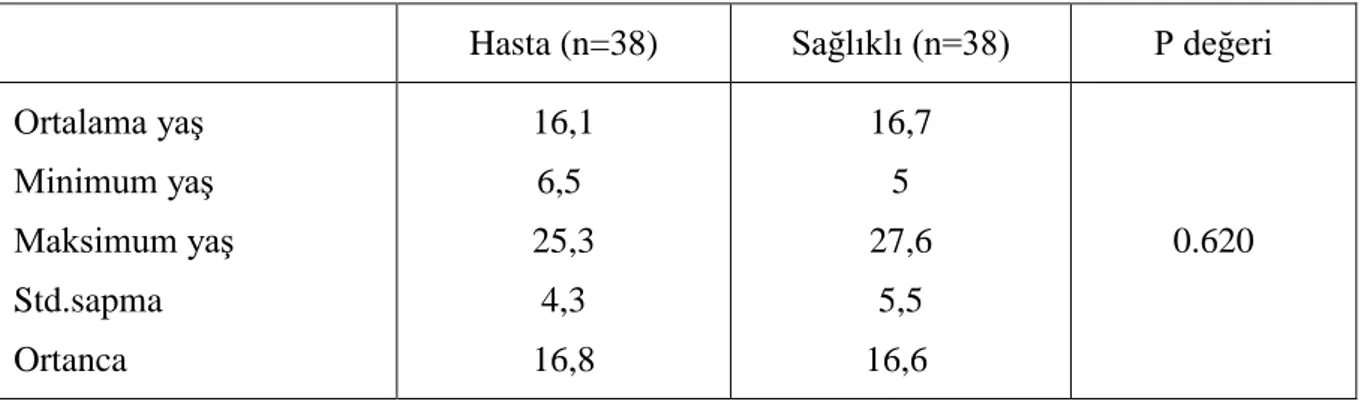
Tablo 12. Olguların Ortalama Yaş Dağılımı ... 31
Tablo 13. Talasemi olguların: Tx yaşı, tanı-tx arası geçe süre, tx-çalışma tarihi arasında geçen süre ve tx öncesinde ferritin düzeyleri ... 31
Tablo 14. HKHN öncesi talasemik değerlendirme ... 32
Tablo 15. Nakil öncesi olguların komplikasyonları ... 32
Tablo 16. Olguların HKHN Özellikleri ... 33
Tablo 17. HKHN sonrası sonrası gelişen komplikasyonlar ... 34
Tablo 18. Olguların eğitim durumunun dağılımı ... 35
Tablo 19. Ebeveynlerin eğitim ve çalışma durumlarının dağılımı ... 35
Tablo 20. Ailelerin ve olguların kişi başına düşen gelirlerin dağılımı... 37
Tablo 21. 18 yaş üstü olguların çalışma ve eğitim durumları ... 37
VI
Tablo 23. 18 yaş altı hasta ve kontrol grubunda, çocuk ve ebeveyn formları ile hayat kalitesinin değerlendirilmesi ... 38 Tablo 24. 5-7 yaş arası hasta ve kontrol grubunda, çocuk ve ebeveyn formları ile
hayat kalitesinin değerlendirilmesi ... 39 Tablo 25. 8-12 yaş arası hasta ve kontrol grubunda, çocuk ve ebeveyn formları ile
hayat kalitesinin değerlendirilmesi ... 40 Tablo 26. 13-18 yaş arası hasta ve kontrol grubunda, çocuk ve ebeveyn formları ile
hayat kalitesinin değerlendirilmesi ... 41 Tablo 27. 18 yaş üstü hasta ve kontrol grubunda hayat kalitesinin karşılaştırılması ... 42 Tablo 28. Tüm yaşlarda, hasta ve sağlıklı olguların toplam yaşam kalite puanlarının
karşılaştırılması. ... 42 Tablo 29. Olguların hayat kalitesini etkileyebilecek risk faktörlerinin
VII
KISALTMALAR
Hb : Hemoglobin Hct : Hemotokrit
MCV : Orta Korpuskuler Hacim
MHC : Ortalama Hemoglobin Konsantrasyonu
MCHC : Ortalama Korpuskuler Hemoglobin Konsantrasyonu RDW : Eritrositlerin Dağılım Genişliği
MRG : Magnit Rezonans Görüntüleme KCFT : Karaciğer Fonksiyon Tstleri ALT : Alanin Aminotransferaz AST : Aspartat Aminotransferaz HLA : İnsan Lökosit Antijeni
HKHN : Hematopoetik Kök Hücre Nakli GVHH : Graft Versus Host Hastalığı AGVHH : Akut Graft Versus Host Hastalığı KGVHH : Kronik Graft Versus Host Hastalığı G CSF : Granülosit Koloni Stimulan Faktör VOD : Venooklüziv Hastalık
TAM : Trombositer Mikroanjiopatik Anemi GİS : Gastrointestinal Sistem
IL : İnterlökin
Gy : Gray
TNF : Tümör Nekrozis Faktör IFN γ : İnterferon gama
ATG : Antitimosit Globulin TBI : Tüm Vücut Işınlama
VIII
SFT : Solunum Fonksiyon Testi CMV : Sitamegalovirüs
VZV : Varisella Zoster Virüs Tx : Transplantasyon MSD : Uyumlu Kardeş Donör MUD : Uyumlu Akrabadışı Donör MRD : Uyumlu Akraba Donör
IX
ÖZET
HEMATOPOETİK KÖK HÜCRE TRANSPLANTASYONU YAPILAN TALASEMİ MAJÖR TANILI HASTALARDA
UZUN DÖNEM YAŞAM KALİTESİNİN DEĞERLENDİRİLMESİ
Giriş: : Beta-Talasemi major, otozomal resesif geçişli, hemoglobin yapısında yer alan β globin zincirlerinin doğumsal olarak sentezinin azalması veya sentezlenememesi sonucu ortaya çıkan, hemolitik anemi ile karakterize, hemoglobinopati grubu bir hastalıktır.
Olguların klasik tedavisi; 3-4 haftada bir kan transfüzyonu ile aneminin düzeltilmesi ve inefektif eritropoez ve kan transfüzyonları sonucu vücutta biriken demirin etkili bir şekilde vücuttan uzaklaştırılmasına dayanır. Talasemi majör hastalarında hem anemi, hem de vücutta ve özellikle dokularda biriken demir, zamanla organ komplikasyonlarına sebep olmakta, yaşam süresini sınırlamaktadır. Başlıca komplikasyonlar kalp, karaciğer, pankreas ve endokrin sistem ile ilgilidir. Günümüzde, iyi-güvenli transfüzyon ve etkili şelasyon tedavisi ile bu olguların yaşam süreleri belirgin uzamış, komplikasyonların gelişimi geciktirilmiştir. Bununla birlikte, yaşam kaliteleri bozuk ve maksimum yaşam süreleri sınırlıdır.
Hematopoetik kök hücre nakli halen mevcut tek iyileştirici seçenektir. Ancak kök hücre nakli kendisi de kısa ve uzun dönemde mortalite ve morbidite riski taşımaktadır. Dünyadaki mevcut deneyim, talasemi majorlu hastaların % 90'ından fazlasının şu anda hematopoetik kök hücre transplantasyonu sonucu hayatta kaldığını ve hastalıksız sağkalımın % 80 civarında olduğunu göstermektedir.
Kök hücre nakli; olguların talasemisiz yaşamalarına olanak tanımaktadır, ancak uzun dönemde yaşam kalitelerine etkisi konusunda yapılan çalışma ve bilgi sınırlıdır.
Amaç: Çalışmamızın amacı çocukluk çağında beta talasemi major tanısı ile allojenik hematopoetik kök hücre nakli yapılan olgularda, nakil sonrası uzun dönemde yaşam kalitesini belirlemek, standart ölçeklerle belirlenen fiziksel ve psikososyal yaşam kalitesi sonuçlarını aynı yaş grubundaki sağlıklı çocukların (aile içi ya da dışı sağlıklı akranlar) sonuçlarıyla karşılaştırmaktır. İkincik amaç; uzun dönem yaşam kalitesini etkileyen tedavi ile ilgili faktörleri belirlemektir.
X
Hipotez: Talasemi major nedeniyle çocukluk çağında HKHN yapılan olguların uzun dömende yaşam kaliteleri sağlıklı akranlarına benzer olmaktadır.
Metod: Çalışmaya Ege Üniversitesi Tıp Fakültesi Pediatrik Kök Hücre Transplantasyon Ünitesi’nde, kurulduğu Ağustos 1998 ile– Ağustos 2017 tarihleri arasında talasemi major nedeniyle HKHN yapılmış ve en az 2 yıldır talasemisiz yaşayan, 5 yaş üzeri 38 olgu alındı.
Kontrol grubu olarak benzer yaş grubundaki sağlıklı çocuklar (hastaların kardeşleri ya da akut sorunlarla genel pediatri polikliniğine başvurmuş çocuklar ve kardeşleri) ve genç, sağlıklı erişkinler (tıp fakültesi öğrencileri) seçildi.
Yaşam kalitesinin değerlendirilmesi için evrensel yaşam kalite ölçekleri: 5-18 yaş arasındaki çocuklarda, Türkçe olarak geçerlilik ve güvenirliği gösterilmiş- PedsQl 4.0 (PediatricQuality of Life Inventory (PedsQLTM) Version 4.0) anket formu , 18 yaş üstü olgularda ise ‘Dünya Sağlık Örgütü Yaşam Kalite Ölçeği-Kısa formu (WHOQOL-BREF)’ kullanıldı. ‘Pediyatrik Yaşam Kalitesi Soru Formu’ gereği anket, 18 yaş altı olgularda hem çocuk, hem de ebeveyne uygulandı ve bedensel, duygusal, toplumsal ve okul ilgili işlevsellik sorgulandı. WHOQOL-BREF ölçeğinde ise fiziksel, psikolojik, sosyal ve çevresel işlevler olgunun kendisi tarafından yanıtlandı.
Ayrıca yaşam kalitesine etkisi olabileceği gözönüne alınarak; 18 yaş altı olguların ve ebeveynlerinin eğitim ve iş durumları, 18 yaş üstündeki olguların ise kendilerinin eğitim ve çalışma durumları ayrı anket formları ile değerlendirildi.
Bulgular: Çalışmaya alınan 38 hasta olgunun 16 sı kız (%42.1) ve 22 si erkek (%57.9) olup ortalama yaş 16.1± 4.3 ( min 6.5, max 25.3) yıl idi. Bu olguların; transplant yaşı ortalama 8.7 ± 3.4 yıl, transplantasyon tarihinden çalışmaya alınma tarihine kadar geçen süre ortalama 7.3±3.2 yıl (min 2 – max 11.6 yıl ) idi. Kontrol grubuna seçilen 38 sağlıklı olgunun 21 i kız (%55.3) ve 17 si erkek (%44.7) olup, ortalama yaş 16.7 ±5.5 ( min 5, max 27.6 yaş) yıl idi.
Çalışmada yer alan hem sağlıklı hem de hasta grubu kendi içinde 4 ayrı yaş grubuna ayrıldı; hem hasta hem de sağlıklı grubun her birinde 5-7 yaş arası 2 olgu (%2.6) , 8-12 yaş arası 5 olgu (%6.6), 13-18 yaş arası 18 olgu (%23.7) ve 18 yaş üzeri 13 olgu (%17.1) yer almaktaydı.
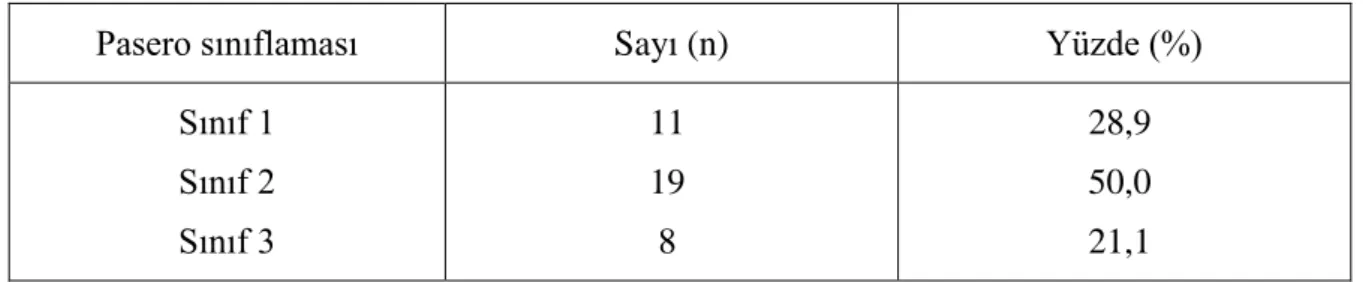
Hastaların, transplantasyon öncesi bakılan ferritin değerleri ortalama 2105±1124 ( min 368, max 5470) ng/dl idi ve Pesaro kriterlerine göre risk sınıflandırılması yapıldığında, 38 olgunun %21’i Class 3 olarak değerlendirildi. Olguların HKHN öncesi talasemik
XI
komplikasyonları incelendiğinde; karaciğer disfonksiyonları 16 olguda (%42.1), endokrin komplikasyonlar 7 olguda (%18.4), kardiyak komplikasyonlar ise bir olguda (%2.6) saptandı.
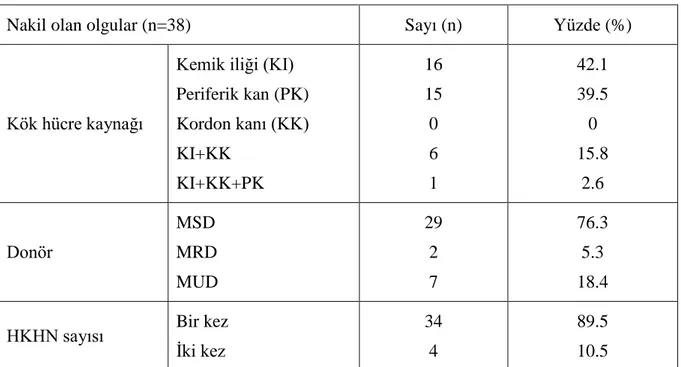
HKHN sırasında donör, 29 hastada MSD - uyumlu kardeş (%76.3), 2 olguda MRD- uyumlu akraba (%5.3), ve 7 olguda MUD - uyumlu akraba dışı donör (%18.4) ‘dür. Kök hücre kaynağı olarak 16 olguda (%42.1) kemik iliği, 15 olguda (%39.5) periferik kan, 6 olguda (%15.8) kemik iliği ve birlikte kordon kanı, 1 olguda (%2.6) kemik iliği+ kordon kanı+periferik kan kullanılmıştır. Otuzsekiz olgudan 4’üne graft yetmezliği nedeniyle 2. kez aynı vericiden HKHN yapılmıştır.
HKHN sonrası gelişen komplikasyonlar değerlendirildiğinde; sadece bir olguda derece 3-4 akut GVHH görülürken, 5 olguda KGVHH ( 2 olguda sınırlı, 3 olguda yaygın) , 17 olguda ( %44.7) endokrin komplikasyonlar görüldü.
Çalışmaya alınan 18 yaş altı tüm olguların ebeveynlerinin eğitim ve iş durumları değerlendirildiğinde hasta ve sağlıklı grup arasında istatiksel olarak anlamlı fark bulunmuştur; hasta çocukların annelerinin % 60’ı ilkokul mezunu iken, sağlıklı kontrol olgularında annelerin % 52’ si yüksekokul-üniversite olduğu, hasta çocukların annelerinin %76’ sı ev hanımı iken, sağlıklı olguların annelerinin % 52’ sinin memur olarak çalıştığı görüldü (p<0.001). Ayrıca hasta çocukların babalarının % 60’ ı ilkokul mezunu iken, sağlıklı olguların babalarının % 52 si yüksekokul-üniversite mezunu, hasta olguların babalarının %56’ sı işçi olarak çalışırken, sağlıklı olguların babalarının % 56’ sı esnaf-tüccar-yönetici olduğu görüldü.
18 yaş üzerindeki hasta ve kontrol grubunda eğitim ve bir işte çalışma oranları 2 grup arasında farklı bulundu (p=0.04). Gerek üniversite düzeyinde eğitim ve mezuniyet, gerekse de bir işte çalışma oranı kontrol grubunda daha fazlaydı.
Bu Çalışmada; Tüm yaş gruplarında; tüm hasta ve kontrol grubunun toplam yaşam kalite puanlamalarına baktığımızda; hasta ve sağlıklı grup arasında istatiksel olarak anlamlı fark bulunmadı.
18 yaş üstü erişkin olguların, hasta ve kontrol grubu arasında yaşam kalitesi açısından tüm alanlarda istatiksel olarak anlamlı farkı olmadığı görüldü.
Çalışmada; 18 yaş altı olguların yaşam kalitesinin değerlendirilmesinde, çocukların doldurduğu anket verilerinde okul faaliyetlerinde, ebeveynlerin doldurduğu anket verilerinde ise bedensel işlevsellik ve toplam yaşam kalitesi puanlarında sağlıklı grup lehine istatiksel olarak anlamlı farklar görüldü (p=0.41, p=0.003, p=0.009).
XII
18 yaş altı olgular yaş gruplarına göre incelendiğinde; 5-7 yaş grubundaki yaşam kalitesi karşılaştırması az hasta sayısı (toplam 2 hasta 2 kontrol grubu) nedeniyle yapılamadı, ancak puanlara baktığımızda çocuk anketlerinde okul faaliyetinde, ebeveyn anketinde ise bedensel, toplumsal, okul faaliyeti ve toplam puanlarda hastalarda sağlıklı olgulara göre daha düşük puanlar bulunmuştur.
8-12 yaş grubunda ise, çocuk verilerine göre – bedensel, duygusal, okul ve toplam puanda; ebeveyn verilerine göre ise tüm puanlarda sağlıklı çocuk lehine istatiksel olarak anlamlı fark görülmektedir ( p=0.008, p=0.031, 0.029, p=0.003, p=0.009, p=0.015, p=0.025, p=0.013, p=0.005).
13-18 yaş grubu olguların yaşam kalitesi değerlendirildiğinde, tüm alanlarda istatiksel olarak anlamlı fark olmadığı görüldü.
HKHN yapılmış olguların yaşam kalitesine etki eden faktörler değerlendirildiğinde; donor tipi, Pesaro risk gruplaması, K GVHD, endokrin ve diğer gelişen komplikasyonların toplam kalite puanlarını ortalamasını anlamlı olarak etkilemediği görüldü.
Tartışma: Çalışmamızda tüm yaş gruplarındaki hastaların, kontrol grubundaki akranlarıyla toplam yaşam kaliteleri puanları arasında fark olmadığı görüldü. Ancak yaş gruplarına göre karşılaştırmalar yapıldığında; 18 yaş üzeri genel yaşam kalite ölçek puanları arasında fark bulunmazken, 18 yaş altı grupta farklı yaş grupları arasında farklar olduğu görüldü.
Çalışmada 18 yaş altı tüm olguların yaşam kalitesinin değerlendirilmesi sonucunda çocuk verilerinde okul faaliyetinde, ebeveyn verilerinde ise bedensel işlevsellik ve toplam yaşam kalitesi puanlarında sağlıklı grup lehine istatiksel olarak anlamlı farklar görüldü (p=0.41, p=0.003, p=0.009). 18 yaş altı hasta ve sağlıklı olguları 3 alt gruba ayırarak yaşam kalitesini değerlendirdiğimizde bu farkı belirgin olarak 8-12 yaş arasındaki olguların yarattığı izlendi. Daha küşük yaş grubunda (5-7 yaş arası) olgu sayısının sınırlı olması (2 hasta, 2 kontrol grubu var) yorumu güçleştirmektedir. 8-12 yaş grubunda olguları değerlendirdiğimizde ise çocuk verilerine göre – sağlığım ve faaliyetlerim, duygularım, okul ve toplam puanda; ebeveyn verilerine göre ise tüm puanlarda sağlıklı çocuk lehine istatiksel olarak anlamlı fark görülmektedir. Yani özellikle 8-12 yaş arası okul çocukları bedensel enerjileri daha düşük, okula adaptasyonları daha az ve duygusal olarak daha kırılgan oldukları görüldü. Literatüre baktığımızda; bu yaş grubu talasemik çocuklarda HKHN ile yaşam kalitesinin arttığı görülmektedir (47). Sağlıklı çocuklarla yapılan karşılaştırma ise bu yaş grubu için yoktur.
XIII
13-18 yaş olguların yaşam kalitesi değerledirildiğinde hem çocuk , hem de ebeveyn verilerinde istatiksel olarak anlamlı fark olmadığı görüldü.
Çalışmamızda 18 yaş üzerindeki hasta ve kontrol grubunda eğitim ve bir işte çalışma oranları 2 grup arasında farklı bulundu. Gerek üniversite düzeyinde eğitim ve mezuniyet, gerekse de bir işte çalışma oranı kontrol grubunda daha fazlaydı. Bu durum hastalardaki yaşam kalitesinin bir ölçeği ve daha düşük yaşam kalitesi olarak yorumlanabilir. Ancak bu yorumu yaparken kontrol grubunun hastalarla aynı sosyal gruptan seçilmediğini ve sadece 7 tanesinin hasta ailelerinin bir bireyi olduğunu gözönüne almamız gerekmektedir. 18 yaş altındaki çalışmaya alınan sağlıklı olguların ailelerinin de hasta ailelerine göre daha eğitimli ve ekonomik gelirlerinin daha iyi olduğunu, kardeş sayısı açısından hasta grubundaki ailelerin daha kalabalık olduğu görmekteyiz. Bu durum bu çalışmanın zayıf yönüdür.
Literatürde talasemik olgularla HKHN yapılmış olguların yaşam kalitelerini karşılaştıran çalışmalar görülmektedir. Ancak HKHN yapılmış olguları sağlıklı akranlarla karşılaştıran çalışma sayısı çok daha azdır. Bu çalışmalarda da yaşam kalitesi erişkin yaş grubunda karşılaştırılmış ve HKHN yapılan talasemilerin yaşam kalitelerinin çok yönüyle sağlıklı bireylere eş olduğu gösterilmiştir. Bizim çalışmamızda da tüm yaş gruplarını birarada değerlendirildiğinde ve 13 yaş üzerinde sağlıklı bireylerle fark saptanmamış sadece küçük yaş grubu (8-12 yaş arası) farklı bulunmuştur. Bu yaş grubunda sağlıklı akranlarla karşılaştırma literatürde saptanmamıştır. Ancak olgu sayılarının sınırlı olması (5 hasta vs 5 kontrol) bu konuda yorum yapmayı güçleştirmektedir. Bu yaş grubundaki hasta çocuklarda ciddi bir tedavi sonrası okula ve normal yaşama adaptasyon daha zor olmuş olabilir yada bu çocukların ebeveynlerinin daha az eğitimli ve daha düşük SES’e sahip olması onlar için bir dezavantaj yaratmış olabilir.
Literatürdeki çalışmalar özellikle KGVHD’nin yaşam kalitesini olumsuz etkilediğini göstermektedir. Bizim çalışmamızda 3’ü ağır 5 KGVHD olgusu mevcuttur ve bu olguların özellikle 2’sinde yaşam kalite puanları daha düşüktür, HKHN yapılmış olguların yaşam kalitesine etki eden faktörler değerlendirildiğinde pesaro riski, donor tipi, K GVHD varlığı ve uzun dönem organ komplikasyonları açısından anlamlı fark bulunmamıştır.
Sonuç olarak; bizim çalışmamızda çocukluk çağında değişik donorlerden talasemi major tanısıyla HKHN yapılan olgularda yaşam kalitesi sağlıklı akranlarına benzer olarak bulunmuştur.
XIV
SUMMARY
ASSESSMENT OF LONG-TERM QUALITY OF LIFE IN PATIENTS WITH THALASSEMIA MAJOR DIAGNOSIS FOLLOWING HEMATOPOETIC STEM CELL TRANSPLANTATION
Introduction: Beta-thalassemia major is a hemoglobinopathy group characterized by hemolytic anemia which is caused by congenital synthesis of β globin chains in autosomal recessive inheritance.
The classical treatment of the cases is based on correcting anemia by blood transfusion every 3-4 weeks and effective removal of iron accumulated in the body as a result of ineffective erythropoiesis and blood transfusions. In thalassemia major patients, both anemia and iron accumulated in the body and especially in tissues cause organ complications over time and shorten the life span. The main complications are related to the heart, liver, pancreas and endocrine system. Nowadays, the survival of these cases has been prolonged and the development of complications has been delayed with good-safe transfusion and effective chelation therapy. However, quality of life is impaired and maximum life expectancy is limited.
Hematopoietic stem cell transplantation is the only curative option currently available. However, stem cell transplantation itself carries a risk of mortality and morbidity in the short and long term. Current experience in the world shows that more than 90% of patients with thalassemia major currently survive hematopoietic stem cell transplantation and disease-free survival is around 80%.
Stem cell transplantation allows individuals to live without thalassemia, but studies and information on the long-term impact on the quality of life are limited.
Objective: The aim of this study was to determine the long-term quality of life in patients with allogeneic hematopoietic stem cell transplantation with beta-thalassemia major in childhood and to compare the results of physical and psychosocial quality of life with standard scales to healthy children in the same age group (healthy peers within or outside the
XV
family). The secondary objective is to identify treatment-related factors that affect long-term quality of life.
Hypothesis: Long-term quality of life in patients with HSCT in childhood due to thalassemia major is similar to healthy peers.
Metods: The study included 38 patients over 5 years of age who had undergone HSCT for thalassemia major between 2005 and August 2017 in Ege University Faculty of Medicine, Pediatric Stem Cell Transplantation Unit.
Healthy children in the same age group (siblings of patients or children and siblings who applied to the general pediatric outpatient clinic with acute problems) and young healthy adults were selected as the control group.
The Universal quality of life scale (PediatricQuality of Life Inventory (PedsQLTM) Version 4.0), in which its validity and reliability has been proved in Turkish, were used to assess the quality of life in children aged 5-18 years. The World Health Organization Quality of Life Instrument, Short Form (WHOQOL-BREF) also was used in patients over 18 years of age. In accordance with The Pediatric Quality of Life Inventory, the questionnaire was administered to both the child and the parent in cases under the age of 18, and physical, emotional, social and school-related functionality was questioned. In the WHOQOL-BREF scale, physical, psychological, social and environmental functions were answered by the patient himself/herself.
In addition, education and work status of the patients under the age of 18 and their parents; their education and working status of the patients over the age of 18 were evaluated with separate questionnaires considering that it may have an impact on the quality of life.
Results: A total of the 38 patients included in the study, 16 were female (42.1%) and 22 were male (57.9%) with a mean age of 16.1 ± 4.3 (min 6.5, max 25.3) years. These cases; The mean age of transplantation was 8.7 ± 3.4 years and the mean time from transplantation to study was 7.3 ± 3.2 years (min 2 - max 11.6 years). The total of the 38 healthy cases selected to the control group, 21 were female (55.3%) and 17 were male (44.7%). The mean age was 16.7 ± 5.5 (min 5, max 27.6 years).
XVI
Both healthy and patient groups were divided into 4 age groups; 2 patients (2.6%) between 5-7 years, 5 patients (6.6%) between 8-12 years (6.6%), 18 patients (23.7%) between 13-18 years and 13 patients above 18 years of age in both patient and healthy groups (17.1%).
The mean ferritin levels of the patients before transplantation were 2105 ± 1124 (min 368, max 5470) ng / dl, and when risk classification was made according to Pesaro criteria, 21% of 38 patients were evaluated as Class 3. When thalassemic complications were examined before HSCT; liver dysfunction was detected in 16 cases (42.1%), endocrine complications in 7 cases (18.4%) and cardiac complications in one case (2.6%).
During HSCT, the donor was MSD-compatible sibling (76.3%) in 29 patients, MRD-compatible relative (5.3%) in 2 patients, and MUD-MRD-compatible non-relative donor (18.4%) in 7 patients. Bone marrow was used in 16 cases (42.1%), peripheral blood in 15 cases (39.5%), bone marrow and cord blood in 6 cases (15.8%), bone marrow + cord blood + peripheral blood in 1 case (2.6%). Four of the 38 patients underwent HSCT for the second time because of graft failure.
When the complications after HSCT were evaluated; Grade 3-4 acute GVHD was seen in only one case, CFSV was limited in 5 cases (limited in 2 cases, common in 3 cases) and endocrine complications in 17 cases (44.7%).
When the educational and occupational status of the parents of all patients under 18 years of age were evaluated, a statistically significant difference was found between the patient and the healthy group; As 60% of the mothers of the sick children were primary school graduates, 52% of the mothers were college-university in healthy control cases, 76% of the mothers of the sick children were housewives, and 52% of the mothers of the healthy cases worked as civil servants (p <0.001 ). In addition, as 60% of the fathers of the sick children were primary school graduates, 52% of the fathers of the healthy cases were college-university graduates, 56% of the fathers of the sick cases worked as workers, and 56% of the fathers of healthy cases were tradesmen-merchant-manager.
The rates of education and employment in the patient and control groups over 18 years of age were different between the two groups (p = 0.04). Both university-level education and graduation, and the rate of working in a job was higher in the control group.
In this study, when we examined the total quality of life scores of all patients and control groups in all age groups, no statistically significant difference was found between patient and healthy group.
XVII
There was no statistically significant difference in quality of life between the patient and control groups in adult cases over 18 years of age.
In this study, The survey data filled by children in school activities, there was a significant difference in the quality of life of the patients under the age of 18, while in the questionnaire filled in by the parents, there were statistically significant differences in physical functionality and total quality of life scores in favor of the healthy group. (p = 0.41, p = 0.003, p = 0.009).
When the patients under the age of 18 were examined according to the age groups, the comparison of the quality of life in the 5-7 age group could not be made because of the small number of patients (2 control groups in total 2 patients). However, when we examined the scores, it was observed that patients scored lower in school activities than healthy cases in the child questionnaires, whereas low scores in participation in physical, social and school activities were observed in the parent questionnaire..
In the 8-12 age group, there are significant differences in physical, emotional, school and total scores according to the data of the child, and this difference was observed in favor of the healthy child in all scores according to the parental data. (p = 0.008, p = 0.031, 0.029, p = 0.003, p = 0.009, p = 0.015, p = 0.025, p = 0.013, p = 0.005).
The quality of life of the 13-18 age group was evaluated, it was seen that there was no statistically significant difference in all areas, while the factors affecting the quality of life of HSCN cases were evaluated, it was seen that donor type, Pesaro risk grouping, K GVHD, endocrine and other developing complications did not significantly affect the mean of total quality scores.
Discussion: In our study, no difference was found between peers and total quality of life scores of the patients in all age groups. However, when comparisons are made according to age groups; While there was no difference between the general quality of life scale scores above the age of 18, there were differences between the different age groups in the age group below 18 years.
In the study, as a result of evaluating the quality of life of all cases under the age of 18, statistically significant differences were found in favor of the healthy group in terms of physical functioning and total quality of life scores in the child data and in the parental data (p = 0.41, p = 0.003, p = 0.009). The limited number of cases (2 patients, 2 control groups) in the
XVIII
younger age group (5-7 years old) makes interpretation difficult. When we evaluate the cases in the 8-12 age group, there are significant differences in health and activities, emotions, school, and total score according to child data. However, these differences become different according to parental data in favor of healthy children in all scores.
In other words, it was observed that school children between 8-12 years of age had lower physical energy, less adaptation to school and less emotionally fragile. When the literature is examined, it is seen that quality of life is increased with HSCT in thalassemic children of the same age group (47). The comparison with healthy children is not available for this age group.
When the quality of life of 13-18 age group was evaluated, it was seen that there was no statistically significant difference in both child and parent data.
In our study, the rates of education and employment in the patient and control groups over 18 years of age were found to be different between the two groups. Both university-level education and graduation, and the rate of working in a job was higher in the control group. This can be interpreted as a measure of quality of life and lower quality of life in patients. However, while making this interpretation, we need to consider that the control group is not selected from the same social group as the patients and only 7 of them are a member of the patient families We observed that the families of the healthy cases under the age of 18 who were included in the study were more educated and had a better income than the patients' families. In addition, we see that the families in the patient group are more crowded in terms of the number of siblings. This is the weakness of this study.
In the literature, there are studies comparing the quality of life of thalassemic and HSCN cases. However, there are fewer studies comparing HSHN cases with healthy peers. (44) (57In these two studies, the quality of life was compared in the adult age group and it was shown that thalassemias undergoing HSCT were very similar in terms of quality of life to healthy individuals. In our study, when all age groups were evaluated together, no difference was found between healthy individuals over the age of 13 and only the younger age group (8-12 years) was found to be different. Comparison with healthy peers in this age group has not been found in the literature. However, the limited number of cases (5 patients vs 5 controls) makes it difficult to comment on this issue. Adolescent children in this age group may have been more difficult to adapt to school and normal life after a serious treatment, or their parents may be less educated and have lower SES, which may be disadvantageous.
XIX
Studies in the literature show that CSVHD especially affects the quality of life negatively (51). In our study, there were 5 cases of CSVHD, 3 of which were severe, and 2 of them had lower quality of life scores. No significant difference was found in terms of pesaro risk, donor type, presence of GVHD and long-term organ complications. As a result; In our study, quality of life was found to be similar to healthy peers in patients who underwent HSCT for thalassemia major from different donors in childhood.
Key words: Beta thalassemia major, Hematopoietic Stem Cell Transplantation, Quality of Life
1
1.GİRİŞ ve AMAÇ
Araştırmanın giriş ve gerekçesi: Talasemi major, hemoglobin yapısında yer alan globin zincirlerinin doğumsal olarak sentezinin azalması veya sentezlenememesi ile karakterize, otozomal resesif geçişli hemoglobinopati grubu hastalıktır. Oluşan anormal hemoglolin aşırı hemoliz ve birlikte inefektif eritropoezis’e sebep olmaktadır. Talasemi, akraba evliliğinin sık olduğu ülkemizde, halen önemli ve önlenebilir bir toplum sağlığı sorunudur.
Olguların klasik tedavisi; 3-4 haftada bir kan transfüzyonu ile aneminin düzeltilmesi ve inefektif eritropoez ve kan transfüzyonları sonucu vücutta biriken demirin etkili bir şekilde vücuttan uzaklaştırılmasına dayanır. Hem anemi, hem de vücutta ve özellikle dokularda biriken demir, talasemi majör hastalarında, zamanla organ komplikasyonlarına sebep olmaktadır. Başlıca komplikasyonlar kalp, karaciğer ve pankreas dahil endokrin sistem ile ilgilidir. İyi-güvenli transfüzyon ve etkili şelasyon tedavisi ile günümüzde komplikasyonların gelişimi ancak geciktirilebilmektedir.
Günümüzde talasemi majorda tek küratif tedavi allojenik kök hücre naklidir. Ancak kök hücre nakli kendisi de kısa ve uzun dönemde mortalite ve morbidite riski taşımaktadır. Kök hücre nakli; olguların talasemisiz yaşamalarına olanak tanımaktadır ve amaç yaşam kalitesini üst düzeye taşımaktır. Kök hücre nakli yapılan talasemi majör olgularının uzun dönemde yaşam kalitesi konusunda yeterli çalışma ve yeterli bilgi yoktur.
Çalışmamızın amacı; çocukluk yaş grubunda kök hücre nakli yapılan talasemi majör olgularının uzun dönemde yaşam kalitesini değerlendirmektir.
Araştırmanın primer amacı:
1. Çocukluk çağında beta talasemi major tanısı ile allojenik hematopoetik kök hücre nakli yapılan olgularda, nakil sonrası uzun dönemde fiziksel ve psikososyal yaşam kalitesini belirlemektir
Sekonder amaçlar:
2. Talasemi majör tanısıyla allojenik kök hücre nakli yapılan olgularda standart ölçeklerle belirlenen fiziksel ve psikososyal yaşam kalitesi sonuçlarını aynı yaş grubundaki sağlıklı çocukların (aile içi ya da dışı sağlıklı akranlar) sonuçlarıyla karşılaştırmak,
2
2.GENEL BİLGİLER
2.1.Talasemi 2.1.1. Tanım
Talasemi ve orak hücre hastalığı dahil kalıtsal hemoglobin bozuklukları dünya çapında en yaygın görülen monogenik hastalıklardır (1).
Talasemiler hemoglobin (Hb) zincirlerinden birinin veya birkaçının hatalı sentezlenmesinin yol açtığı hipokrom mikrositer anemi ile karakterize heterojen bir grup hastalıktır. Talasemiler otozomal resesif geçiş göstermektedir. Talasemiler Hb α, β, γ, δ olarak klasifiye edilen hemoglobin zincirlerinin bir ve ya birkaçının sentezlenememesi sonucu oluşmakta ve bu tanımlamaya göre alfa zincir sentez azlığı alfa talasemi, beta zincir sentez azlığı beta talasemi olarak adlandırılmaktadır (2)(3).
Dünyanın birçok bölgesi için, son veriler ( her ne kadar güvenilir veriler hala eksik olsa da), dünya nüfusunun yaklaşık % 7'sinin bir hemoglobin bozukluğunun taşıyıcısı olduğunu ve her yıl 300.000-500.000 çocuğun bu hastalıkların ağır homozigot durumu ile doğduğunu gösteriyor (4).
Alfa talasemi Güney Çin, Malezya ve Tayland'da oldukça yaygındır. Beta talasemi Afrika'da oldukça yaygındır. Nüfustaki tahmini heterozigotluk oranı Afrika'da yaklaşık % 13, Asya'da % 4 ve ABD'de % 2 ve ülkemiz genelinde %2,3 olan β talasemi taşıyıcılık oranı Antalya yöresinde yapılan bir araştırmada %10’un üzerinde bulunmuştur. Ülkemizde yaklaşık 4.500 ne yakın talasemili hasta olduğu tahmin edilmektedir. (5)(6).
2.1.2. Talasemide sınıflama
Alfa talasemiler aglobin zincirlerinin sentezlenememesi sonucu oluşmaktadır. Alfa -globin genleri 16. Kromozomun kısa kolunum telomerik ucunda bulunur. Alfa talasemi genellikle bir veya iki a-globin genini içeren büyük DNA fragmanlarının silinmesinden kaynaklanır (4).
Beta talasemilerde - her kromozomda bir tane olmakla iki beta globin geni vardır. Beta talasemi, bu genlerin birindeki veya her ikisindeki mutasyonlardan kaynaklanır. Hastalığın şiddeti normal beta globin üretiminin miktarı ile ilişkilidir. Mutasyonlar, azalmış ekspresyon (beta +) veya ekspresyonun tamamen yokluğuna (beta 0) sebep olabilmektedir. Beta globin ekspresyonu süt çocuğu döneminde başladığı için (gama globin fetal ve erken bebek
3
hemoglobinlerinde kullanılan beta benzeri zincirdir), talasemi fenotipi genellikle yaşamın ilk yılında ortaya çıkmaya başlar (tipik sunum yaşı- dört ila altı ay) ve bundan dolayı beta talasemi yenidoğanlarda bulgu vermez (4).
Beta talasemiler 4 farklı klinik olarak karşımıza çıkabilmektedir. 1. Sessiz taşıyıcı: hematolojik olarak normal,
2. Talasemi minör (taşıyıcı, heterozigot):hafif mipokrom mikrositer anemi, 3. Talasemi intermedia (hasta, homozigot):transfüzyon ihtiyacı fazla olmayan,
4. Talasemi majör (hasta, homozigot): transfüzyon bağımlı olarak sınıflandırılmakta (2)(3). Sessiz taşıyıcı- globin sentezinde orta derecede azalma, hematolojik olarak normal, kan transfüzyonu gerektirmeyen, orta derece anemi ile karakterizedir.
Beta talasemi taşıyıcılığı- 3 farklı tipte prezende eder: 1. Yüksek A2 ile olan ve en fazla görülen tip,
2. Yüksek A2 ve yüksek F ile beta talasemi taşıyıcılığı ve 3. Normal A2 ile olan beta talasemi taşıyıcılığı (1)(3).
Beta talasemi taşıyıcılığı tanısı konarken– demir eksikliği anemisi, alfa talasemi taşıyıcılığı ve kronik hastalık anemisi ayırıcı tanısının yapılması gerekmektedir. Beta talasemi taşıyıcılığında tedaviye gerek yoktur ancak taşıyıcılık yönünden aileye genetik danışmanlık önerilmelidir (3).
Beta talasemi intermedia – homozigot talasemi olmasına rağmen klinik bulgular beta talasemi majör kadar ağır değildir. Olgularda Hb değerleri genellikle 6-10 g/dl düzeyindedir, Hemotokrit (Hct), eritrosir sayısı ve eritrosit indekslerinde (MCV, MHC, MCHC) azalma, RDW de artış, periferik yaymada: hipokromi, mikrositoz, anizositoz, poikilositoz, hemoglobin elektroforezinde: HbA %10-20, HbF %70-80, HbA2 normal sınırlarda izlenmektedir. Beta talasemi intermediada de büyüme-gelişme, kemik değişiklikleri ve splenomegali gelişimi açısından izlenmelidir. Tedavi olarak: kan transfüzyonu, demir şelasyonu ve ileri vakalarda gerektiğinde splenektomi yapılmaktadır (3) (4) (7).
Beta talasemi major - talasemi major'ın klinik prezentasyonu 6 ile 24 ay arasında gerçekleşir. Etkilenen bebekler gelişemez. Beslenme sorunları, ishal, sinirlilik, tekrarlayan ateş atakları, dalak ve karaciğer büyümesi görülebilir. Kapsamlı tetkik ve tedavisi yapılamayan veya kötü transfüzyonunun yapıldığı bazı gelişmekte olan ülkelerde, talasemi major'un klinik tabloları; büyüme geriliği, solukluk, sarılık, zayıf kas sistemi, hepatosplenomegali, bacak ülseri, genu vakgum görülmektedir. Ekstramedüller hematopoez
4
ve kemik iliğinin genişlemesinden kaynaklanan iskelet değişiklikleri: bacakların uzun kemiklerindeki deformasyonları ve tipik kraniyofasiyal değişiklikler talasemi majorlu olgularda tipiktir (3) (8).
Minimum Hb konsantrasyonunu 9.5 ila 10.5 g / dL tutan düzenli bir transfüzyon programı başlatıldığında, büyüme ve gelişme 10 ila 12 yıla kadar normal olma eğiliminde olmaktadır. Transfüzyon yapılan hastalarda aşırı demir yüklenmesine bağlı komplikasyonlar gelişebilir. Çocuklarda aşırı demir yüklenmesi komplikasyonları birçok organ sistemini etkilemektedir. Aşırı demir yüküne bağlı komplikasyonlar arasında kardiyak komplikasyonlar (dilate miyokardiyopati veya nadiren aritmi), karaciğer komplikasyonları (fibrozis, hepatit ve siroz), endokrin komplikasyonlar (diyabetes mellitus, hipogonadizm, hipotiroidi, hipoparatiroidi), büyüme gelişme ğeriliği yer almaktadır. Diğer komplikasyonlar hipersplenizm, kronik hepatit, HIV enfeksiyonu, venöz tromboz ve osteoporozdur. Karaciğerde viral enfeksiyonu ve aşırı demir yükü olan hastalarda hepatoselüler karsinom riski artar. Miyokardiyal siderozun neden olduğu kardiyak hastalık beta-talasemi hastalarında yaşamı sınırlayan en önemli demir komplikasyonudur. Aslında, kalp komplikasyonları beta-talasemi majörlü hastaların% 71'inde ölümlerin nedenidir (9).
2.1.3. Talasemide tedavi prensipleri
Beta talasemi majorda temel tedavi ve izlem prensipleri aşağıdaki gibidir: Güncel tedaviler:
a. eritrosit transfüzyonu, b. demir şelasyon tedavisi, c. splenektomi,
d. komplikasyonların izlem ve tedavisi.
Küratif tedavi- Kök Hücre Transplantasyonu (3)(10).
a) Eritrosit transfüzyonu –büyüme-gelişme geriliği olan, ağır anemi ile ilişkili klinik bulguları olan, ilerleyici splenomegalisi olan olgularda transfüzyon yeterli olması daha da önemlidir. Eritrosit transfüzyonu kuralları:
Transfüzyon öncesinde hemoglobin düzeyi 9-10 g/dl olmalı, Transfüzyon sonrası Hb 14-15 g/dl’i aşmamalı,
Transfüzyon hızı 4-5 ml/kg/saati geçmemeli, Tranfüze edilen kan miktarı 10-15 cc/kg,
5
Transfüzyon sıklığı 3-4 hafta aralıklarla olmalıdır (4) (10).
b) Demir şelasyon tedavisi- düzenli transfüzyon rejimi uygulanan hastalarda aşırı demir yüküne bağlı olarak : hipogonadizm , hipotiroidi ,hipoparatiroidi , diyabet , karaciğer fibrozu ve kalp fonksiyon bozukluğu gibi klinik bulgular ortaya çıkmaktadır (4).
Şelasyon tedavinin esas amacı vücutta biriken demirin önlenmesi, mevcut demir birikiminin azaltılması ve böylece demir birikimine bağlı komplikasyonların önlenmesidir. Olgularda düzenli transfüzyonun ilk yılını doldurduğunda ve ya 12-15 transfüzyon sonrası, serum ferritini 1000 ng/ml düzeyine ulaştığında demir şelasyon tedavisinin başlanması önerilmektedir. Bu tedavinin amacı – serum ferritini 500-1000 ng/ml, karaciğer demirini 3-5 mg Fe/g kuru karaciğer ağırlığı ve kardiyak MRI (T2) düzeyini >20 ms üzerinde tutmayı hedeflemektedir. Talasemili olgularda kullanılan demir şelatör seçenekleri:
Desferrioksamine (Desferal, DFO) Deferiprone (Ferriprox, DFP) Deferasiroks (Exjade, DFX) (4)(10).
c) Splenektomi - Talassemili olgularda şiddetli hemoliz, dalağın ilerleyici aşırı aktivitesi ile sonuçlanır, bu da aneminin şiddetini arttırır ve sonuç olarak transfüzyon gereksinimlerini arttırır.
Endikasyonları :
Eritrosit süspansiyonu kalitesi yeterliyken, transfüzyon öncesi hemoglobin değerini 9-9.5 g/dl arasında tutmak için gerekli yıllık kan tüketimi 200-220 ml/kg eritrosit süspansiyonu üzerinde olan olgularda
Hipersplenizm bulguları olan olgularda splenektomi önerilmektedir.
Splenektomi çocuklada ciddi enfeksiyon riski sebebiyle 2 yaşından küçük çocuklarda yapılmamaktadır. Postplenektomi sepsisi ile en sık ilişkili olan patojenler kapsüllenmiş organizmalar, özellikle: Streptococcus pneumoniae (asplenik hastalarda kanıtlanmış bakteriyel enfeksiyonların % 75'inden fazlasını oluşturur) , Haemophilus influenzae Neisseria meningitidis suçlanmaktadır. Bundan dolayı splenektomiden yaklaşık 1 ay öncesinde kapsüllü bakterilerden korunma amacıyla pnömokok, hemofilius influenza, meningokok aşıları yapılmalı ve splenektomi sonrasında olgulara antibiyotik profilasi tedavisi başlanmalıdır (3)(4)(11).
6
d) Komlikasyonların izlem ve tedavisi:
1. Kardiyak komplikasyon- kalp kası hücrelerinde demir birimine bağlı komplikasyonlar ( kalp yetmezliği, kardiyomiyopatiler, aritmiler) beta talasemili olgularda en sık ölüm nedenidir. Bundan dolayı olgular belli aralıklarla EKG ve EKO ile değerlendirilmelidir. Kardiyak demir yükünü değerlendirmek için MRG tetkiki yapılmalıdır (12) .
2. Endokrin komplikasyonlar - Aşırı demir yüküne bağlı endokrinopati, talasemili hastalarda kronik transfüzyon tedavisinin en sık bildirilen komplikasyonudur. Komplikasyonlar arasında : anemi , şelasyon tedavisi sonucu demir birikimi sonucunda olgularda büyüme gelişme geriliği, puberta tarda, hipogonadizm, ekstremite kırıkları, daibetes mellitus, hipotiroidi , hipoparatiroidi görülmekte. Bundan dolayı olgularda tanıdan itibaren her 3 ayda bir olgularda bu komplikasyonların izlemi açısından boy, kilo, tiroid fonksiyon testleri, kemik dansitometresi, puberta evrelemesi, OGTT yapılmalı ve sonucuna göre tedavi planlanmalıdır (4)(13).
3. Hepatik komplikasyonlar – transfüzyona bağlı viral enfeksiyonlar ( hepatit B ve C) ve karaciğerde demir yüküne bağlı olarak gelişen karaciğer hasarı – fibrozis, siroz ve karsinom ortaya çıkmaktadır. Bu komplikasyonların izlemi açısından olguda her 3 ayda bir KCFT , Total Bilirubin , Direk Bilirubin bakılmalıdır . Karaciğerde biriken demir miktarını ölçmek için karaciğer MRG yapılmalıdır(3)(4).
2.2.Hematopoetik Kök Hücre Nakli (HKHN) 2.2.1.Tanım
Hematopoetik kök hücre nakli, günümüzde hemoglobinopatiler için mevcut tek kürativ tedavidir. Son yıllarda komplikasyonların önlenmesi ve tedavisinde tıbbi yönetimdeki gelişmeler nedeniyle Talasemi majorlu bireylerin tedavisinde başarılar elde edilmiştir ve yaşam süreleri belirgin uzamıştır. Bununla birlikte, hematopoetik kök hücre nakli hala mevcut tek iyileştirici seçenektir. Dünyadaki mevcut deneyim,talasemi majorlu hastaların % 90'ından fazlasının şu anda hematopoetik kök hücre transplantasyonu sonucu hayatta kaldığını ve hastalıksız sağkalımın % 80 civarında olduğunu göstermektedir (6) (14).
Tüm talasemi major tanılı hastalarda aileleleri tanı aldıktan sonra kök hücre nakli tedavisi için bilgilendirilmeli , isterlerse doku grupları (HLA) araştırılmalı , donörü olma olasılığı açısından değerlendirilmelidir. HLA uygun kardeş donör bulma şansı % 25 gibidir. Günümüzde en sık uygulanan, HLA-uygun kardeşten alınan kök hücre transplantasyonudur.
7
Nakil öncesinde olgular hepatomegali, karaciğer biyopsisinde fibrozis varlığı, şelasyon tedavisine uyuma göre Class I, II ve III olarak sınıflandırılmıştır ( Pesaro sınıflaması) .
Tablo 1. Talassemik değerlendirme (3).
Sınıflama Şelasyon Hepatomegali Fibrozis
Class 1 Class 2 Class 3 Düzenli Düzenli/düzensiz Düzensiz Yok +/- Var Yok +/- Var
Hematopoetik kök hücre nakli (HKHN), hematopoetik progenitör hücrelerin bir donörden (örn. allojenik, otolog) ve herhangi bir kaynak (örn. kemik iliği, periferik kan, göbek kordon kanı) kullanarak alınması ve hastaya kemik iliğini yeniden oluşturmak için verilmesini ifade eder (15).
Talasemi major tanılı olgularda allojenik hematopoetik kök hücre nakli uygulanmaktadır.
Allojeneik hematopoetik kök hücre nakli.
Allojeneik HKHN, bir akrabadan (HLA uyumlu , haploidentik veya uyumsuz ) ve ya akraba dışı (gönüllü veya umbilikal kordon donörü) donörden toplanan hematopoetik progenitör hücrelerin kullanımı anlamına gelir . HKHN'yi hemen takip eden pansitopeni süresi, otolog HKHN'den daha uzundur ve immünosupresif ajanlar GVHH'ye karşı profilaksi ve tedavi için yaygın olarak kullanılır.Allojenik HKHT sonrası sağkalım destekleyici bakımdaki teknolojik ilerlemeler, histokompalibilite testlerindeki gelişmeler, daha güvenilir hazırlık rejimleri, daha etkili GVHH profilaksisi ve fırsatçı enfeksiyonların daha iyi yönetimi ile birlikte gittikçe artmaktadır (16).
8
2.2.2.Kök hücre kaynakları 2.2.2.1. Kemik iliği
Kemik İliği Hematopoetik kök hücre nakli için klasik olarak kabul edilen kök hücre kaynağıdır. Kemik iliği genel anestezi altında iliak kemikten toplanır. Toplanan kemik iliğinin yeterliliğinde belirleyici olan çekirdekli hücre sayısıdır. Başarılı bir engraftman için önerilen çekirdekli hücre sayısı alıcı vücut ağırlığı başına 2-4x10⁸’dir. Kemik iliğindeki kök hücre miktarını arttırmaya yönelik granülosit koloni stimülan faktör (GCSF) uygulanması, erişkin vericilerde bildirilmekle birlikte, çocuk vericiler ile ilgili veriler çok sınırlıdır(17).
2.2.2.2. Periferik kan kök hücresi
Pek çok merkezde Otolog transplantasyon için progenitör hücrelerin kaynağı olan periferik kan, şimdi birçok hematolojik hastalık için standart tedavi yöntemidir. Son yıllarda erişkin vericilerden yapılan akraba ve hatta akraba dışı nakillerde artan sıklıkta kullanılmaktadır. Bu uygulamanın en büyük avantajı nötrofil ve trombosit engrafman sürelerinin beklenenden daha kısa olması ve hastanede kalış süresinin, transfüzyon gereksiniminin ve enfeksiyon sıklığının daha az olmasıdır. Bütün bunlar nakil maliyetini azaltan faktörlerdir. Ancak ürün toplama işleminin kendisi özellikle uygun venöz yol sağlama ile problemler, donöre kateter takma gereksinimi, mobilizasyonla ilgili kullanılan ajanların (GCSF) yan etkileri, artmış GVHH riski mutlaka göz ardı edilmemesi gereken temel faktörlerdir (18).
2.2.2.3. Göbek kordon kanı
Göbek kordon kanı mükemmel bir hematopoetik kök hücre kaynağıdır. Başarılı göbek kordonu kan naklinin ana belirleyicileri, nakil ve HLA eşleşmesi için mevcut olan hücrelerin sayısıdır. Hızlı kullanılabilirlik ve GVHH'ye neden olma sıklığının azaltılması avantajlarına sahiptir. Bu, sınırlı HLA uyumu ile greft nakli için izin verir ve böylece donör havuzunu artırır. Doğum sırasında toplanan göbek kordon kanında nispeten yüksek sayıda hematopoetik kök hücre vardır. Bu hücreler kordon kanı bankalarında işlenebilir ve dondurulabilir. Dünya çapında bu tür bankalarda birkaç yüz bin kordon kanı ünitesinin bulunduğu tahmin edilmektedir. Bir alıcıya HLA-eşleşmesinin ardından, kordon kanı birimi, transplant merkezine en az gecikmeyle taşınabilir ve kullanılabilir (19).
9
2.2.3. Hazırlanma rejimi
Talasemili major tanılı olgularda miyeloablatif hazırlık rejimi uygulanmaktadır.
2.2.3.1. Miyeloablatif hazırlama rejimi
Konvansiyonel miyeloablatif rejimi ile amaç konakta olan T lenfositleri elimine ederek immun aracılı başarısızlığı ortadan kaldırmak ve altta yatan hastalığa ait olan rezidüel hücreleri eradike etmektir. Malign hematolojik hastalıkların tedavisinde HKHT prosedürlerinin uygulamaya girmesinden sonra birçok miyeloablatif hazırlama rejimi geliştirilmiştir ve bunların çoğu halen kullanılmaktadır. Ancak yüksek doz TBI veya kemoterapi, organlarda hasara ve GVHH'na predispozisyon oluşturabilecek sitokin fırtınasına neden olarak organ ve dokulara önemli zararlara neden olmaktadır. GVHH’nı önlemek için nakil sonrası dönemde tekli ve ya kombinasyon halinde immunsupresif ajanlar kullanılmaktadır (20) (21)
2.2.3.2. Nonmiyeloablatif hazırlama rejimi
Nonmiyeloablatif kök hücre transplantasyonu, yaşlı olgular ve ciddi komorbiditeleri olanlar için yeni bir nakil yöntemi olarak ortaya çıkmıştır. Miyeloablatif rejimlerle karşılaştırıldığında, nonmiyeloablatif hazırlama rejimleri akciğerler, karaciğer, böbrekler için daha az toksik etkisi görülmekte , daha az enfeksiyona neden olmakta, daha az kan transfüzyonuna gerek duymakta ve eşlik eden komorbiditesi olan yaşlı hastalarda bile daha düşük bir ölüm oranıyla ilişkilendirilmektedir (21).
Tablo 2. Yaygın Kullanılan Ajanlara ve Kombinasyonlara Göre Miyeloablatif ve Miyeloablatif Rejim Örneği (22).
Miyeloablatif TBI ≥5 Gy tek doz veya ≥8 Gy fraksiyonlu
Busulfan > 8 mg / kg oral veya intravenöz eşdeğeri
Nonmiyeloablatif
TBI≤ 2 Gy ± purin analoğu
Fludarabin + Siklofosfamid ± ATG Fludarabin + Sitarabin + İdarubisin Kladribin + Sitarabin
10
2.2.4. Engraftman
Kök hücre transplantasyonunda başarı göstergelerinden birisi verilen hematopoietik kök hücrelerin hastanın kemik iliğine yerleşerek yaşam için gerekli olan kan hücrelerini dışarıdan yardım gerekmeyecek şekilde sentezlemeye başlaması ve devam ettirmesidir. Kök hücre infüzyonundan sonraki zaman diliminde gerçekleşen bu sürece engraftman adı verilmektedir. Engraftman myeloid seri, lenfoid seri ve trombosit engraftmanı olarak alt gruplara ayrılmaktadır (23).
Engraftman kriterleri
Nötrofil engraftmanı- 3 gün süre ile nötrofil sayısı > 0.5 x 10 9
/ L seyreder ise myeloid engraftmanın bu 3 günün 1. gününde olduğu gösterir.
Trombosit engraftmanı – 7 gün trombosit transfüzyonuna gerek olmadan trombosit sayısının ardışık 3 gün >20 000/mm³ ve bunu izleyen günlerde >50 000mm³ olduğu ilk gün (24)(25).
Transplantasyon sonrası acil yönetim: hematopoetik destek, enfeksiyon profilaksisi ve greft versus-host hastalığının (GVHH) önlenmesine yöneliktir. Otolog ve allojenik hematopoetik hücre nakli peri-transplantasyon döneminde nötropeni, anemi ve trombositopeni ile ilişkilidir. Miyelosupresyonun derecesi ve hematopoetik iyileşme süresi, hazırlık rejimi ve greft kaynağı dahil olmak üzere birçok faktöre göre farklılık gösterir. Kan ürünü nakli ve hematopoetik büyüme faktörleri, nakil bakımının temel komponentleridir .Kan ürünü desteği genellikle hematopoetik hücre nakli (HKHN) öncesinde, sırasında ve sonrasında gereklidir (26).
2.2.5. Kök hücre nakli sonrası gelişen komplikasyonlar
Her yıl yaklaşık 50.000 kişiye dünya çapında hematopoetik hücre nakli yapılmaktadır. Transplantasyon tekniklerindeki ve destekleyici bakım uygulamalarındaki ilerlemeler, kök hücre nakli yapılan olguların yaşam oranlarında iyileşmelere yol açmıştır. Hastalar nakil sonrası uzun süre hayatta kaldıklarından, nakil öncesi, nakil sırasında ve nakil sonrası risklerle ilgili geç komplikasyonlar açısından risk altındadırlar. Bu komplikasyonlar ciddi morbiditeye neden olabilir, yaşam kalitesini boza bilir ve kök hücre alıcılarında geç mortaliteye katkıda bulunabilir (27)
11
Yüksek doz kemoterapi ve/veya radyoterapinin yanı sıra, GVHH’nin profilaksisi veya tedavisi için immünosüpresif ilaç uygulamaları, nakil sonrası bir çok komplikasyonun oluşmasına neden olmaktadır (28).
2.2.5.1. Erken dönem komplikasyonlar
Erken komplikasyonlar nakil sonraki ilk 100 gün içinde ortaya çıkan komplikasyonlar olarak tanımlanabilir. Bu dönemde epitelyal ve endotelyal hücreler ilaçlara bağlı toksisiteye maruz kalırlar. Özellikle kemoterapiye bağlı gasto-intestinal sistemde oluşan mukozal hasar sonucu oluşan oral mukozit, bulantı, kusma, ishal meydana gelmektedir. Üriner sistem epitelinde toksisite sonucu hemorajik sistit; endotel hasarı sonucunda venooklüziv hastalık (VOD), trombotik mikroanjiyopatik anemi (TAM) ya da engraftman sendromu gibi çok farklı klinik tablolar ortaya çıkabilmektedir (28).
2.2.5.1.1.Akut graft versus host hastalığı (AGHH)
GVHH, önemli morbidite ve mortalite ile ilişkili olan allojenik HKHT’nun majör komplikasyonudur. Aktif donör T hücreleri, hazırlama rejiminin tetiklediği inflamatuar bir kaskadtan sonra konakçı epitelyal hücrelerine zarar verir. GVHH için risk faktörleri - hastanın yaşı, kök hücre kaynağı, hazırlama rejimi ve GVHH profilaksine bağlıdır . Akut GVHH’nın etkilediği üç ana doku deri, karaciğer ve gastro-intestinal sistem hücreleridir. Klinik olarak dermatit, karın ağrısı±diare, bulantı, kusma, hepatit (bilirübin veya karaciğer enzim yüksekliği) geliştiğinde tanıdan şüphelenilmelidir (29).
Akut GVHH klinik bulguları tipik olarak nakilden sonra ilk 100 gün içinde ortaya çıkar, Kronik GVHH bulguları tipik olarak nakilden 100 gün sonra ortaya çıkar (30).
Akut GVHH öncelikle cildi (GVHD'li hastaların% 81'i), gastrointestinal sistemi (% 54) ve karaciğeri (% 50) etkiler. Deri lezyonları genellikle ilk ortaya çıkar. Etkilenen hastalarda tipik olarak, boynun ve omuzların etrafında başlayan ve sıklıkla avuç içine kadar yayılan makülopapüler döküntüler oluşmaktadır (30).
12
Tablo 3. Akut GVHH’nın klinik belirtileri (30). Organ Klinik belirtiler
Deri Eritematöz makülopapüler döküntü Bütün vücut yüzeyine yayılabilir. Kaşıntılı ve/veya ağrılı olabilir Ağır durumlarda büller oluşabilir
Karaciğer Sarılıkla birlikte olan veya olmayan kolestaz.
Kolestaz enzimlerindeki bozukluk, transaminazlardakinden daha fazladır GİS İştahsızlık, bulantı, kusma
İshal; yeşil renkli ve sulu
Ağır durumlarda dışkıda taze kan görüle bilir Bazen paralitik ileus görülmekte
Tablo 4. Akut GVHH’nda evrelendirme (29). EVRE (STAGE) DERİ KARACİĞER (Bilirübin) BAĞIRSAK (Gaita çıkışı/gün) 0 GVHH döküntüsü yok <2 mg/dl <500 ml/gün ve ya kalıcı mide bulantısı
1 Vücut yüzey alanının <%25
makulopapüler döküntü 2-3 mg/dl 500-999 ml/gün
2 Vücut yüzey alanının %25-50
sinde makulopapüler döküntü 3.1-6 mg/dl 1000-1500 ml/gün
3 Vücut yüzey alanının >%50
makulopapüler döküntü 6.1-15 mg/dl >1500 ml/gün
4 Yaygın eritrodermi+büll >15 mg/dl >2000 ml/gün ve ya ileus tablosu
Derece (Grade)
1 Stage 1-2 Yok Yok
2 Stage 3 veya Stage 1 veya Stage 1
3 - Stage 2-3 veya Stage 2-4
13
A GVHD tedavisi: Grade I aGVHH tedavisinde sadece topikal steroidler yeterli olmaktadır. Daha ileri dereceli olgularda sistemik tedavi gerekmekte ve temel tedavi olarak 1-2 hafta süreyle 2 mg/kg/gün metilprednizalon kullanılmaktadır. Standart dozda başlanan steroid tedavisine yanıt alınamaması : tedavi başladıktan sonra 5 gün içerisinde hastalığın progresyon göstermesi ve ya 1-2 hafta içerisinde tam yanıt alınamaması ve ya ilk steroid dozunun azaltılmasından sonra hastalığın nüks etmesi ileri basamak tedaviyi gerektirmektedir .En sık kullanılan 2. Basamak tedaviler arasında T hücrelerini monoklonal antikorlar ve ya ATG yer almaktadır (29) (30).
2.2.5.1.2. Enfeksiyoz komplikasyonlar
Enfeksiyon nedenli komplikasyonlar sitopenisi olan, immun ablasyon ve /ve ya immunsupresyon sebebiyle kök hücre naklinden kısa zaman sonra sık görülmektedir .İmmun sistemin rekonstriksiyonu genellikle 1-1.5 yıl gibi yavaş zaman diliminde oluşmaktadır. Özellikle HLA uyumsuz, göbek kord kanı ve T hücreleri tükenmiş olan graft ile nakli gerçekleşen allojenik alıcılarda , uzun süre immunsupressif tedavi kullananlarda , GVHH olan olgularda immun sistemin yeniden yapılanması daha yavaştır (27).
Bakteriyel, fungal ve viral enfeksiyonlar , gecikmiş immün yeniden yapılandırması olan hastalarda nakilden aylar veya yıllar sonra ortaya çıkabilir. Enfeksiyon riski, nakilden sonraki ilk 1-2 yılda en yüksek olmasına rağmen, uzun süreli immünosüpresif tedavi gerektiren kronik GVHD hastaları gibi bazı allojenik nakil alıcıları için yüksektir. Kronik GVHD hastalarında opsonizasyon bozulmuş ve kapsüllü bakterileri ( N. meningitides, H. influenzae ve S. pneumonia) hızla ilerleyici ve hayatı tehdit edici enfeksiyonlara neden olabilir. Ayrıca, hastalar, altta yatan hastalıklarının tedavisi için splenektomi geçirmiş olabilir veya fonksiyonel olarak GVHH'ye veya splenik ışınlamaya sekonder asplenik olabilir. GVHH için kombine aspleni ve immünsüpresyon riski olan hastalar antibiyotik profilaksisi almalıdır (27).
2.2.5.1.3. VOD
Veno-oklüziv hastalık HKHN'den sonraki ilk haftalarda sık görülen bir komplikasyondur. Küçük intrahepatik venüllerin tıkanması ile karakterizedir ve ilaç toksisitesine bağlı olarak sinüzoid endotel hücrelerinin hasarı sonucu ortaya çıkmakta . Bu tıkanıklık genellikle karaciğer asinüsünün zona 3 de görülmekte. Bu alan, ilaç metabolitlerinin temizlenmesinde ana enzimatik sistemi temsil eden sitokrom P -450 bakımından
14
zengindir. Ancak bu komplikasyon görülen olguların hepsinde hepatik venül tutulumu olmadığından, ‘sinüzoidal obstrüksiyon sendromu (SOS)’ terimi önerilmektedir. VOD başlangıcı en sık HKHN 'den sonraki ilk 30 gün içinde ortaya çıkar ve semptomlar hepatomegali, portal hipertansiyon ve assitlere yol açan hepatik akıştaki azalma ile ilgilidir. VOD multiorgan yetmezliği ile ilişkili olabilir ve trombosit transfüzyonlarına, akciğer tutulumuna (pulmoner infiltratlar ve plevral efüzyon), santral sinir sistemi tutulum semptomlarına (ensefalopati ve koma) ,böbrek ve kalp yetmezliğine neden olabilir.VOD tedavisinde Defibrotide (10 mg / kg ) kullanımının etkili olduğu gösterilmiştir (28).
2.2.5.1.4. Trombotik mikroanjiyopati,
Trombotik mikroanjiyopati, nakil sonrası komplikasyonların en ağır vasküler hasarlarından birini temsil eder. Risk faktörleri arasında siklosporin veya takrolimus kullanımı, dişi cinsiyet, TBI, haploidentik veya akraba dışı donör, GVHH, enfeksiyonlar yer almakta. Trombotik mikroanjiopati (TAM) 'nin klinik bulguları hemolitik anemi, trombositopeni ve böbrek ve / veya merkezi sinir sistemi bozukluğunu içerir. Primer engraftrasyondan sonra eritrosit ve trombosit sayısında bir düşüş meydana geldiğinde TAM'den şüphelenilmelidir (28).
2.2.5.1.5. Hemorajik sistit
Hemorajik sistit, HKHN den sonra görülen yaygın bir morbidite nedenidir. Nakil sonrası erken (ilk 72 saat içinde) veya geç (bir ay sonra) dönemde ortaya çıka bilir. Hazırlama rejimindeki ajanların toksik etkisiyle veya viral bir enfeksiyonun etkisi sonucu ortaya çıkmakta. Erken başlangıçlı hemorajik sistit genellikle siklofosfamid, busulfan ve etoposid gibi kemoterapi ajanlarının katabolizma ürünlerinin neden olduğu mukozal hasarla ilişkilidir. Geç başlangıçlı hemorajik sistit ise genellikle viral enfeksiyonlarla ilşkilidir ve esas olarak insan poliomavirüs tip BK, JK veya adenovirüs suçlanmaktadır. Çocuklarda hemorajik sistit için risk faktörleri sekiz yaşından küçük olmak, donörün akraba dışı olması ve erkek cinsiyettir. Klinik semptomlar arasında enfeksiyon ve ya kanama diyatezi olmadan - mikroskobik hematüri, gross hematüri, dizüri vardır (28).
15
2.2.5.1.6. Graft yetmezliği
Graft yetmezliği HKHN den sonra önemli olan mortalite ve morbidite nedenidir.
Tablo 5. Graft yetmezliği tipleri (31) (32).
Primer graft yetersizliği
Kemik iliği ve periferik kök hücre naklinde 28 gün, kord kanı naklinde 6 hafta nötrofil sayısının ardışık 3 gün ≥ 0.5x109 /L olmaması durumunu kasar.
Sekonder graft yetersizliği
Engrafmanı takip eden zamanda kemik iliğinde hastalığın nüks bulgusu olmaksızın mutlak nötrofil sayısının tekrar < 0.5 x 10 9
olması durumudur
2.2.5.1.7. Engraftment sendromu
Hem otolog hem de allojenik HKHN’nin erken döneminde nötrofil engraftmanının hemen öncesinde ve ya zamanında ortaya çıkan ateş, deri döküntüsü, kardiyojenik olmayan pulmoner ödem ,bazende böbrek yetmezliğinin eşlik ettiği klinik tablo engrafment sendromu olarak tanımlanmakta. Genellikle nötrofil engraftmanının başladığı ilk 3-4 gün içerisinde proinflamatuar sitokinlerin aşırı sentezlenmesi nedeniyle meydana gelir. Multiorgan yetmezliği bulguları engraftmandan bağımsız olarak hazırlama rejimi ile ilişkili yaygın endotelyal hasar, diffüz inflamasyon ve tromboz sebebi ile ortaya çıkmaktadır. Etiyolojisi tam olarak bilinmemekte, ancak multifaktorler suçlanmaktadır. Hazırlama rejimleri sonrası ortaya çıkan sitokinler- IL-1, TNF a , interferon gama epitel ve endotel hücrelerine zarar vermektedir. Engraftman sendronunda klinik bulgular: tanımlanmış enfeksiyon olmadan 38,3°C ve üzerinde ateş, eritrodermik döküntü, diffüz pulmoner infiltrasyon ,hipoksik kardiyojenik olmayan pulmoner ödemdir. Tedavi, birkaç gün boyunca günde 2 mg / kg / gün steroid uygulamasından oluşur. Bu, özellikle erken uygulandığında semptomların çözülmesine yol açar (28) (33).
2.2.5.2. Geç dönem komplikasyonlar
Geç komplikasyonlar ilk 100 günlük nakilden sonra meydana gelir ve bulgular uzun zaman diliminde ortaya çıkar. Nakil sonrası döneminde ortaya çıkan sorunlar, nakil tamamlanmasından sonra çözülmeyen herhangi bir ters olay veya nakil prosedürünün tamamlanmasından sonra ortaya çıkan yeni bir problem olarak tanımlanmaktadır (34).