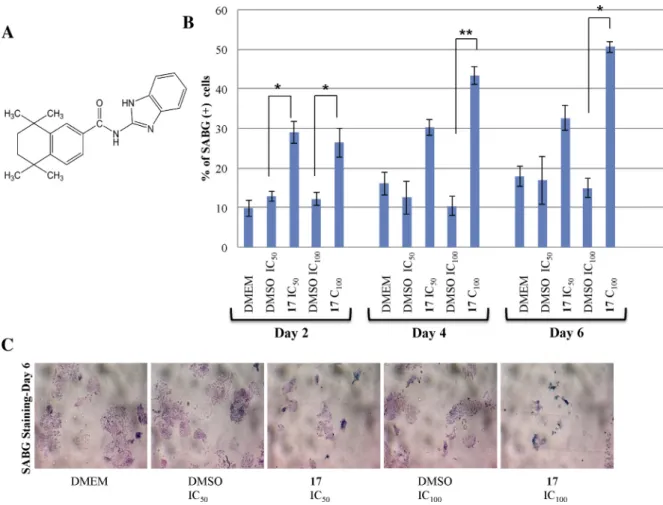
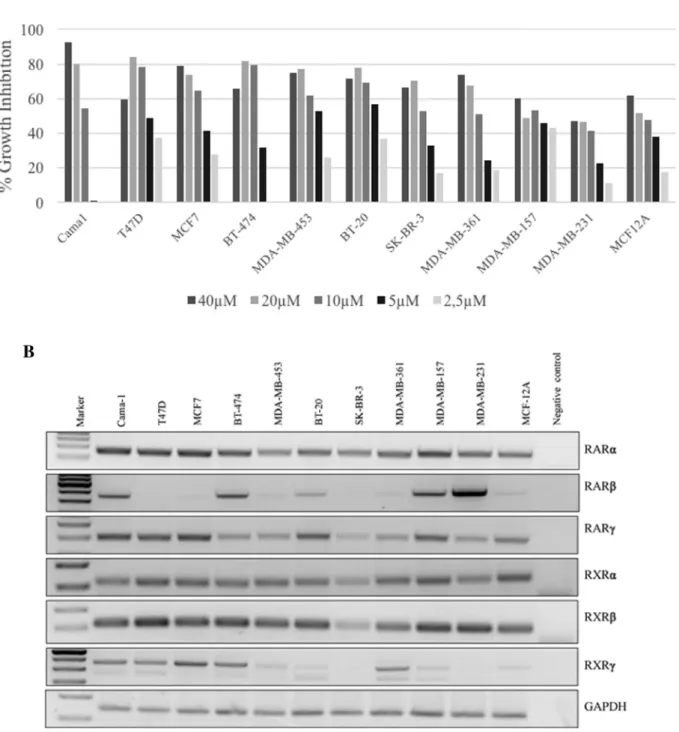
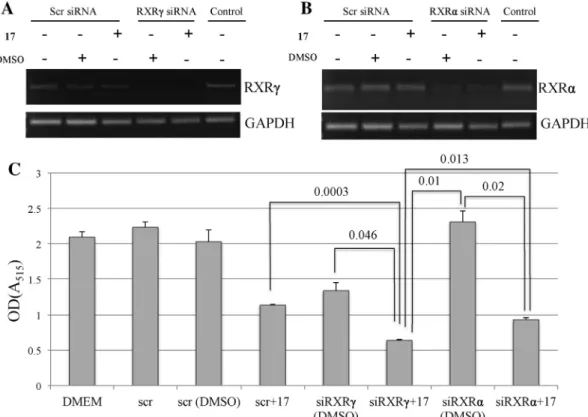
Retinoid N-(1H-benzo[d]imidazol-2-yl)-5,5,8,8-tetramethyl-5,6,7, 8-tetrahydronaphthalene-2-carboxamide induces p21-dependent senescence in breast cancer cells
Tam metin
Şekil
Benzer Belgeler
Türkiye’de Tarım sektöründe faaliyet gösteren tarımsal kooperatifler 1163 Sayılı Kooperatifler Kanunu ve bu Kanuna değiĢik 3476 sayılı kanuna göre faaliyet gösteren
After optimizing the germane flow rate during deposition, the film thickness, and the poling time for maximum peak nonlinearity, we demonstrated a record peak nonlinear coefficient
Finally, we use them to derive a num- ber of results in quantum information theory: from new bounds for quantum random access codes (QRACs) for both real and complex quantum systems
Figure 14: East- and southeast-oriented rooms with OPright were significantly different than the other rooms when the areas of sunlight patches on the total surfaces analyzed at
Following that, Fallout 3 will be presented within the suggested framework of Aarseth composed of three parts, game-world, gameplay & game-structure, while comparing
We will see that under certain circumstances, noise separation can be realized effectively in fractional Fourier domains: Fractional Fourier transforms can be used to separate
These lateral and vertical heterostructures have inhomogeneous magnetic moment configurations due to p−d hybridization; in both sides of the junction, chalcogen atoms have
Gönüllü sade yaşam tarzının bir diğer alt boyutu olan “maddi olmayan hayat”ın içsel geleneksellik değeri ile düşük dereceli pozitif yönlü (r = 0,251) ilişkili
