IDENTIFICATION of NOX SPECIES ADSORBED on COBALT(II)–
SUPPORTED ZIRCONIA and SULFATED ZIRCONIA and INVESTIGATION of THEIR REACTIVITY TOWARD METHANE
A THESIS
SUBMITTED TO THE DEPARTMENT OF CHEMISTRY AND THE INSTITUTE OF ENGINEERING AND SCIENCES OF
BILKENT UNIVERSITY
IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE
OF
MASTER OF SCIENCE
By
AHMET SELİM VAKKASOĞLU
I certify that I have read this thesis and in my opinion it is fully adequate, in scope and in quality, as a thesis of the degree of Master of Science
__________________________________ Assoc. Prof. Margarita Kantcheva (Supervisor)
I certify that I have read this thesis and in my opinion it is fully adequate, in scope and in quality, as a thesis of the degree of Master of Science
__________________________________
Prof. Dr. Şefik Süzer
I certify that I have read this thesis and in my opinion it is fully adequate, in scope and in quality, as a thesis of the degree of Master of Science
_______________________________ Prof. Dr. Yavuz İmamoğlu
I certify that I have read this thesis and in my opinion it is fully adequate, in scope and in quality, as a thesis of the degree of Master of Science
__________________________________ Prof. Dr. Atilla Aydınlı
I certify that I have read this thesis and in my opinion it is fully adequate, in scope and in quality, as a thesis of the degree of Master of Science
__________________________________ Assoc. Prof. Ömer Dağ
Approved for the Institute of Engineering and Sciences
__________________________________ Prof. Dr. Mehmet Baray
ABSTRACT
IDENTIFICATION of NOX SPECIES ADSORBED on COBALT(II)–
SUPPORTED ZIRCONIA and SULFATED ZIRCONIA and INVESTIGATION of THEIR REACTIVITY TOWARD METHANE
AHMET SELİM VAKKASOĞLU M.S. in Chemistry
Supervisor: Assoc. Prof. Margarita Kantcheva July 2002
The development of new catalysts for the selective catalytic reduction (SCR) of
nitrogen oxide (NOx) emissions from both stationary and mobile sources has gained an
increasing interest in the past decade. This is due to the need for replacing the NH3-SCR
process with possibly a CH4-SCR process (methane-selective catalytic reduction) and
demands for improvement of fuel economy in internal combustion engines with low CO2
emissions. However, little progress had been made since the discovery in 1992 of
cobalt(II)-exchanged ZSM5 and ferrierite as effective catalysts for the SCR of NOx with
methane in presence of oxygen. Most of the catalytic systems studied are severely affected by the presence of water vapor and do not show practical activity under actual exhaust conditions, at least below 773 K. The search for highly active and hydro-thermally stable catalysts for reduction of NO with methane in presence of oxygen at low reaction temperatures continues.
The present work is a fundamental study aimed at investigation the possibility of application of solid acids based on sulfated zirconia (active in the isomerization of light
saturated hydrocarbons) as catalysts for CH4-SCR of NO. The activation of light alkanes
in the isomerization process on catalysts such as sulfated zirconia starts with cleavage of the C−H bond. The same process is important in the activation of methane in the SCR of NO. Sulfated zirconia possesses moderately strong surface acidity, weaker than that of
ZSM5. However, its structure is not sensitive to water and the surface sulfate groups are stable up to 850 K.
This work involves FTIR spectroscopic studies on the interaction of methane with the surface of cobalt(II) supported on zirconia and sulfated zirconia and investigations of
the role of preadsorbed NOx species in the process of hydrocarbon activation. The
samples have been prepared by wet impregnation of zirconia and sulfated zirconia using cobalt(II) acetate as a precursor. The catalyst containing 5.3 wt % of cobalt and 4.5 wt% of sulfated ions (CoSZ sample) has been investigated in detail. In order to understand the effect of the sulfate ions on the performance of the cobalt(II)-zirconia system, the same amount of cobalt (5.0 wt %) was introduced into sulfate free-zirconia (CoZ sample). The catalysts have been characterized by XRD and in situ FTIR spectroscopy. The monoclinic structure of zirconia support converts to tetragonal after the deposition of cobalt(II). The sulfated zirconia and the CoZS catalyst have the same phase composition (67 % tetragonal). The application of sulfated zirconia as support causes decrease in the average crystallite size, increase in the surface area and better dispersion of the cobalt ions. The introduction of cobalt(II) to zirconia and sulfated zirconia leads to increase in the basicity of the surface hydroxyls.
The adsorption of NO on both catalysts reveals that there are no exposed
coordinatively unsaturated Zr4+ ions and leads to formation of cobalt(II) dinitrosyls. In
addition, a process of NO disproportionation with the involvement of the surface OH
groups takes place producing anionic nitrosyl (NO–), nitro (NO2−) species and water
molecules. The sulfate groups decrease the reducibility of the Co2+ sites and oxidize
readily the anionic nitrosyls.
On coadsorption of NO and O2 at room temperature on the samples studied, various
kinds of surface nitro-nitrato species are observed differing in the modes of their coordination. The nitro-nitrato structures on the sulfated sample are characterized by a lower thermal stability than that on the CoZ sample. The difference in the thermal
stability of the NOx− (x = 2 and/or 3) parallels their activity toward methane. Most of the
nitrate species on the sulfate-free catalyst do not interact with the hydrocarbon and they
CoSZ catalyst are able to oxidize the methane producing formic acid, formate, carbonate/carboxylate species and water. The presence of sulfate ions in the CoSZ
catalyst affects the reactivity of the adsorbed NOx species and increases the acidity of the
Lewis sites, which facilitates the activation of methane. The NOx-modified CoSZ catalyst
displays higher activity than the NOx-free catalyst, which shows the importance of the
surface nitro-nitrato complexes in the activation of methane as selective reducers of NO. Taking into account the higher hydrothermal stability of sulfated zirconia than that of ZSM5, the materials containing cobalt(II) supported on sulfated zirconia could be
promising as CH4-SCR catalysts.
Keywords: Adsorption of NO and NO/O2; in situ FTIR; cobalt(II) supported on zirconia
ÖZET
KOBALT(II) DEPOLANMIŞ ZİRKONYUM ve ZİRKONYUM SÜLFAT ÜZERİNE
ADSORBE OLMUŞ NOx MOLEKÜLLERİNİN TANIMLANMASI ve METAN ile
TEPKİMESİNİN ARAŞTIRILMASI
AHMET SELİM VAKKASOĞLU Kimya Bölümü Yüksek Lisans
Tez Yöneticisi: Assoc. Prof. Margarita Kantcheva Temmuz 2002
Sabit ve hareketli kaynaklardan yayılan azot oksit (NOx) moleküllerinin seçici
katalitik indirgenmesinde kullanılan katalizörlerde yapılan geliştirme çalışmalarına olan ilgi son yıllarda artmıştır. Bu ilginin iki sebebinden biri, seçici katalitik indirgenme yönteminde kullanılan amonyağın yerine metan kullanımı, diğeri ise içten yanmalı motorlardan yayılan karbondioksit gazının azaltılmasıdır. Bununla birlikte, 1992’de,
oksijenli ortamda metan ile NOx moleküllerinin seçici katalitik indirgenmesinde etkili
katalizör görevi yapan kobalt(II) depolanmış ZSM5 zeolitinin keşfedilmesinden bu yana fazla gelişme sağlanamamıştır. Birçok katalitik sistemin su buharından ciddi şekilde etkilendiği görülmüştür. Ayrıca bu sistemler gerçek atık ortamlarında (<773K) aktifliklerini yitirmişlerdir. Azot oksiti (NO) metan ile oksijenli ortamda, düşük sıcaklıklarda indirgeyecek, yüksek aktiviteye sahip ve hidrotermal kararlılığı olan katalizörlere yönelik çalışmalar devam etmektedir.
Bu çalışmanın amacı, katı asit olan zirkonyum sülfatın (az sayıda karbonu olan doygun hidrokarbonların izomerizasyonunda aktiftir) NO’in metan ile seçici katalitik indirgenmesinde uygulanabilirliğini araştırmaktır. Sözü geçen hidrokarbonların zirkonyum sülfat üzerindeki izomerizasyon tepkimelerinde aktif hale gelmeleri C–H bağının homolitik olarak kırılması ile olur. NO’in seçici katalitik indirgenmesi tepkimesi sırasında metanın aktifleşmesinde aynı sürecin önemi büyüktür. Zirkonyum sülfat,
ZSM5’den zayıf fakat orta kuvvette yüzey asitliğine sahiptir. Bununla birlikte, yapısı suya karşı duyarlı olmadığı gibi sülfat gruplarıda 850 K’e kadar kararlıdır.
Bu çalışma kobalt(II) depolanmış zirkonyum ve zirkonyum sülfat yüzeyleri ile
metanın etkileşmesinin ve yüzeye önceden tutunmuş NOx moleküllerinin hidrokarbon
aktifleşmesindeki rolünün FTIR spektroskopisi ile araştırılmasını içerir. Numuneler zirkonyum ve zirkonyum sülfatın kobalt(II) asetat öncüsü ile ıslak impregnasyon yöntemiyle hazırlanmıştır. Ağırlıkça 5,3% kobalt ve 4,5% sülfat iyonu içeren numune (CoSZ) detaylı olarak incelenmiştir. Sülfat iyonlarının kobalt-zirkonyum sistemi üzerindeki etkisini anlamak için, aynı miktar kobalt (ağırlıkça 5%) zirkonyuma eklenmiştir (CoZ). Katalizörler, XRD ve kendi ortamında FTIR spektroskopisi ile nitelendirilmiştir. Kobalt depolanması monoklinik olan zirkonyumun yapısını tetragonale çevirmiştir. Zirkonyum sülfat ve CoSZ katalizörü aynı faz kompozisyonuna sahiptir (67% tetragonal). Zirkonyum sülfatın destek olarak uygulanması ortalama kristal boyutunda azalmaya, yüzey alanında artışa ve kobalt iyonlarının daha iyi yayılmasına sebep olur. Zirkonyum ve zirkonyum sülfata kobalt(II) eklenmesi yüzey hidroksillerinin bazlığında artışa neden olur.
NO adsorpsiyonu, koordinasyona açık Zr4+ iyonlarının olmadığını ve kobalt(II)
dinitrosillerinin oluştuğunu göstermiştir. Ayrıca NO, yüzey hidroksilleri ile tepkime
vererek anyonik nitrosil (NO–), nitro (NO2–) türleri ve su moleküllerinin oluşmasına
neden olur. Sülfat grupları Co2+ atomlarının indirgenebilirliğini azaltmış ve anyonik
nitrosilleri yükseltgemiştir.
NO ve O2’nin oda sıcaklığında beraber adsorpsiyonu sonucu yüzeyde,
koordinasyonları farklı olan nitro-nitrato türleri gözlenmiştir. Sülfat içeren numunedeki nitro-nitrato yapıları CoZ numunesindekilere oranla daha düşük termal kararlılığa
sahiptir. Bu fark, NOx– (x=2 ve/veya 3)’lerin metana karşı aktifliği ile paraleldir. CoZ
katalizöründeki birçok nitrat türleri hidrokarbon ile tepkime vermeyip, nitro (NO2–)
yapılarına dönüşmüşlerdir. Öte yandan CoSZ katalizöründeki nitro-nitrato türleri metanı yükseltgeyerek, formik asit, format, karbonat/karboksilat türleri ve su oluşumuna sebep
olur. CoSZ katalizöründeki sülfat iyonlarının varlığı, adsorbe olan NOx türlerinin tepkime
NOx adsorbe olmuş CoSZ katalizörü, NOx adsorbe olmamışa oranla daha fazla aktiflik
gösterir. Bu, metanın, NO’in seçici indirgeni olarak aktifleşmesinde yüzeydeki nitro-nitrato yapılarının önemini gösterir. Zirkonyum sülfatın ZSM5’e nispeten daha yüksek hidrotermal kararlılığını hesaba katarak, zirkonyum sülfat destekli kobalt(II) içeren malzemeler, metan ile seçici katalitik indirgeme tepkimesinde katalizör olarak kullanımlarında umut vericidir.
Anahtar Kelimeler: NO ve NO/O2 adsorpsiyonu; kendi ortamında FTIR; kobalt(II)
depolanmış zirkonyum ve zirkonyum sülfat; metan ile seçici katalitik indirgeme; ara ürünler.
ACKNOWLEDGMENT
It is a pleasure for me to express my deepest gratitude to Assoc. Prof. Margarita Kantcheva for her great help and supervision throughout my studies.
I appreciate the moral support by dear friends, Burak Okumuş, Talal Shahwan, Ercan Avcı, Dündar Akarca, Erkan Z. Çiftlikli, Onur Atasoylu, Özlem Demir, Olga Samarskaya, Banu Altıntaş, Burak Birkan, Hüseyin Karakuş, Erkan Köse, A. Çağrı Ateşin, Bayram Erdem, Necmi Bıyıklı, Özgür Karakuzu, Mustafa Keşir, H. Hikmet Erdoğan, Ahmet Günay, İshak Uysal, Ali Canlıer, Ünal Şen, Osman T. Aydaş, Tevfik Emre Aksoy and Twin Brothers.
I would like to thank all present and former members of the Bilkent University Chemistry Department for their help.
TABLE OF CONTENTS
1. INTRODUCTION 1
1.1 Sources of NOx……….1
1.2 Selective Catalytic Reduction of NOx with Ammonia (NH3–SCR)………….2
1.3 SCR of NOx with Methane over Zeolite Catalysts………2
1.4 Selective Catalytic Reduction (SCR) of NOx over Non–zeolitic Oxides with Methane……….4
1.5 Mechanism of the SCR of NO with Methane in Excess Oxygen……….5
1.6 Non–zeolitic Solid Acid Supports for CH4–SCR Catalysts………..6
1.7 Acidity and Reactivity of Modified Zirconias………..7
1.8 Concluding Remarks……..………10
1.9 Identification of Strongly Adsorbed NOx– Species (x=2 or 3) by FTIR Spectroscopy………...11
2. EXPERIMENTAL 15 2.1 Sample preparation……….15
2.2 Surface Area Measurements and X–ray Diffraction………..16
2.3 IR Spectroscopy……….16
2.4 Experimental Setup………16
2.5 Activation of the Samples………..16
2.6 Adsorption of NO and Co–adsorption with O2………..18
2.7 Interaction of CH4 with Catalysts………..18
2.8 Interaction of CH4 with NOx pre–covered catalyst………18
3. RESULTS AND DISCUSSION 19 3.1 Characterization of the Samples……….19
3.1.1 BET Surface Area Measurements and X-ray Diffraction…………...19
3.2 NO and NO/O2 Co–adsorption and Thermal Stability of NOx Species……..25
3.2.1 Adsorption of NO at room temperature on CoZ sample……….25
3.2.2 Coadsorption of NO and O2 on the CoZ Sample and Thermal Stability of the NOx Species Produced.……….….………29
3.2.3 Adsorption of NO at room temperature on CoSZ sample…………..35
3.2.4 Coadsorption of NO and O2 on the CoSZ Sample and Thermal Stability of the NOx Species Produced……….39
3.2.5 Summary of the Results on NO and NO/O2 Adsorption on the Catalysts Studied………..43
3.3 Surface NOx Species toward Methane………45
3.3.1 CoZ Catalyst………...45
3.3.1.1 Blank NOx experiment……….45
3.3.1.2 Interaction of methane with the NOx-precovered CoZ sample…47 3.3.1.3 Blank Experiment With Methane………49
3.3.2 CoSZ Catalyst………50
3.3.2.1 Blank NOx Experiment………50
3.3.2.2 Reactivity of NOx Species Adsorbed on CoSZ catalyst toward Methane………...52
3.3.2.3 Interaction of Methane with the CoSZ Catalyst………..56
4. CONCLUSION 59
LIST OF TABLES
1. Frequencies of Adsorbed NOx Species Observed on Metal Oxides
and Their Corresponding Structure……….13
2. The nominal content of the active components and the notation of
the samples……… ……… …… ………… ……….15
3. Surface areas and loading of the sample studied……….19
4. Assignments of the FTIR Bands Observed during Adsorption
of 0.67 kPa NO and Its Co-adsorption with O2 (NO:O2=1:1) at RT
on CoZ……….33
5. Assignments of the FTIR Bands Observed during Adsorption of
0.67 kPa NO and Its Co-adsorption with O2 (NO:O2=1:1) at RT
LIST OF FIGURES
1 Model proposed by Arata and Hino………..8
2 Model proposed by Kustov et al………...9
3 Model proposed by Adeeva et al………..9
4 IR cell and the furnace………17
5 Vacuum/adsorption apparatus and IR cell………..17
6 Powder X–ray diffraction patterns of the ZrO2, SZ, CoZ, CoSZ samples...21
7 FTIR spectra of the activated catalysts and supports………..23
8 FTIR spectra of adsorbed NO (1.07 kPa) on the CoZ catalyst at room temperature for various times. The spectrum of the activated sample is used as a background reference………..26
9 Results of curve-fitting procedure applied to the FTIR spectra taken after NO adsorption (1.07kPa) on CoZ sample at room temperature. The spectrum of the activated sample is used as a background reference…..28
10 Correlation between the integrated absorbances of the bands at 1909-1913 cm-1 and 1880-1887 cm-1………..29
11 FTIR spectra obtained upon (a) adsorption of NO (0.67kPa) at RT for 5 min, (b) followed by addition of O2 (0.67kPa) for 30 min, and (c) evacuation at RT for 15 min. The spectrum of the activated sample is used as a background reference………...30
12 FTIR spectra taken after heating the catalyst CoZ containing adsorbed NOx species (spectrum 15’ evacuation (RT)) for 15 min in vacuum. The spectrum of the activated sample is used as a background reference………..32
13 FTIR spectra of adsorbed NO (1.07 kPa) on the CoSZ catalyst at room temperature for various times. The spectrum of the activated sample is
used as a background reference………36
14 Results of curve-fitting procedure applied to the FTIR spectra taken after
NO adsorption (1.07kPa) on CoSZ sample at room temperature. The
spectrum of the activated sample is used as a background reference………..37
15 Correlation between the integrated absorbances of the bending mode of
water and asymmetric stretching mode of the cobalt(II) dinitrosyl
at 1795 cm-1……….38
16 FTIR spectra obtained after heating of the CoSZ sample containing preadsorbed
NOx species for 15 min in vacuum at various temperatures.
The spectrum of the activated sample is used as a background reference…...41
17 FTIR spectra of the catalyst CoZ taken after adsorption of NO/O2 mixture (1.33
kPa, NO:O2 = 1:1) at room temperature followed by evacuation for
10 min (a), after heating the closed cell for 30 min at 623 K (b) and 723 K (c). The spectrum of the activated sample is used as a background reference………..46
18 FTIR spectra of the catalyst CoZ taken after adsorption of NO/O2 mixture (1.33
kPa, NO:O2 = 1:1) at room temperature followed by evacuation for
10 min and addition of 6.7 kPa of methane (a) and after heating the closed IR cell for 30 min at 573 K (b), 623 K (c) and 723 K (d). The spectrum of the activated sample is used as a background reference……….48
19 FTIR spectra of the CoZ sample after addition of methane (6.7kPa) at
20 A: FTIR spectra of the catalyst CoSZ taken after adsorption of NO/O2
mixture (1.33 kPa, NO:O2 = 1:1) at room temperature followed by
evacuation for 10 min (a) and after heating the closed IR cell for 30 min at 623 K
B: FTIR subtraction spectrum of the catalyst CoSZ obtained from the spectra shown in Figure 20.A. The spectrum of the activated sample is
used as a background reference………..51
21 A: FTIR spectra of the catalyst CoSZ taken after adsorption of NO/O2
mixture (1.33 kPa, NO:O2 = 1:1) at room temperature followed by
evacuation for 10 min (a); after heating the closed IR cell for 30 min at 623 K followed by cooling to room temperature and addition of 6.7 kPa of methane (b) and subsequent heating the closed IR cell for 30 min at 623 K (c), 673 K (d) and 723 K (e).
B: FTIR subtraction spectra of the catalyst CoSZ obtained from the spectra shown in Figure 21.A. The spectrum of the activated sample is
used as a background reference………...52
22 FTIR spectra of the Co–ZSM5 sample obtained by (a) contact with
NO/O2 mixture (10.67kPa) at room temperature followed by evacuation
for 15 min; (b) addition of methane (2kPa) and subsequent heating the closed IR cell at 433 K for 10 min. The spectrum of the activated sample is used as a background reference………...55
23 FTIR spectra of the CoSZ sample after addition of methane (6.7kPa)
at RT (a); and after heating the closed IR cell for 30 min at 573 K (b); 623 K (c); and 723 K (d). The spectrum of the activated sample is used
24 Comparison of the spectra of the catalyst CoSZ presented in Figure 18,
1. INTRODUCTION
1.1 Sources of NOx
Emission of NOx (NO and NO2) is a major contributor to the acidification of the
atmosphere and soil and to formation of photochemical smog. Major sources of NOx
emissions are combustion of liquid and fossil fuels in the transportation devices, power plants for electricity production, house heating and chemical industry [1].
The main reason for generation of NOx is the high temperature combustion,
which permits oxidation either of nitrogen in the air and/or of nitrogen–containing
components of the fuel to NOx. There are two general approaches to minimize the
concentration of NOx in the waste gases: treating NOx at the source or not to produce
it at all. The former approach is important in the case of flue gases stemming from
nitric acid manufacture and transport devices. If one can avoid the production of NOx,
no post treatment units are necessary. The formation of NOx can be minimized by
more efficient burner designs or addition of more O2 to the fuel [2].
Another technology that has attracted much attention in the last decade (effective for gas turbine application) is the low temperature catalytic combustion
[3-5]. This novel technology has the potential to achieve ultra low emissions of NOx, CO
and unburned hydrocarbons. However, the catalytic combustion for power production has not yet reached commercialization because problems with the long-term performance of the catalyst and technical design of the process are not solved. In addition, lowering of the combustion temperature of the fuel can lead to higher levels
of N2O formation [6], which is a greenhouse gas.
The post-combustion NOx control technologies involve selective non-catalytic
and catalytic methods. The selective non-catalytic reduction (SNCR) implies the addition of reducing agent, such as ammonia, to the effluent gases with conditions of
high temperature and O2 availability [7,8]. The catalytic methods offer lower
operating temperature and the selective catalytic reduction of NOx with ammonia has
1.2 Selective Catalytic Reduction of NOx with Ammonia (NH3–SCR)
In this process, high conversion (>95%) can be achieved at reasonably low temperatures (573 – 673 K). However, many disadvantages still stay in the way of its
application in sectors other than industry. A catalyst reactor system and NH3 injection
unit into the flue gas are needed. This requires sophisticated equipment. Another
disadvantage is the transportation and the storage of anhydrous NH3. Therefore,
capital and operation costs are high. In addition, there is a possibility for slip of
unreacted NH3, which is environmentally objectionable [11]. The high costs of the
NH3–SCR makes this process applicable to larger installations. Smaller and mobile
applications need an alternative DeNOx technology. In this respect, use of methane as
reducing agent is attractive because methane is abundant and easy to handle. Especially with stationary engines operating with natural gas, the selective catalytic
reduction (SCR) of NOx with CH4 would be very cost-effective, as methane is not
corrosive as ammonia and it is a major component of the exhaust gas. Use of methane as reducing agent is also viable for mobile lean-burn engines powered by natural gas. This would be especially favourable, since vehicles fueled with natural gas do not
produce soot or sulfur compounds and emit less CO2 (due to higher H/C ratio for
CH4). In addition, an advantage in favour of CH4 over hydrocarbons with higher
carbon numbers, is the much lower production of N2O [12], another greenhouse gas.
In conclusion, the application of CH4 for control of the NOx emissions by
post-injection after the combustion process [13] according to the equation (1):
2NO + CH4 + O2 N2 + CO2 + 2 H2O (1)
would be a beneficial process for large and small installations.
1.3 SCR of NOx with Methane over Zeolite Catalysts
In the middle of 1980s, Iwamoto et al. [14,15] have discovered the Cu-ZSM5 catalyst for the SCR of nitrogen oxides with hydrocarbons in an excess of oxygen.
This catalyst is non-selective catalyst in the presence of CH4. Later (in 1992), Li and
Armor [16] reported that the Co-ZSM5 catalyst can be particularly effective for the
SCR of NOx with CH4. Other zeolite-based catalysts such as cobalt-exchanged
toward nitrogen. Other transition metal ions (Mn2+– and Ni2+–) exchanged ZSM5 and ferrierites [21] are reported due to their high activity in the reduction of NO with methane but their selectivity is lower than that of Co – FER [22].
Among the platinum group metals, Pd–exchanged ZSM5 catalyst possesses high activity for the SCR with methane in dry conditions [23]. Introducing water
vapour into the feed stream causes considerable reduction of the conversion of NOx.
The addition of Rh to the Pd-ZSM5 system results in high activity in presence of
water [24]. However, selectivity of this catalyst to N2O increases with increase in the
oxygen content in the stream [25,26]. As a result, the palladium–exchanged zeolite system lacks of sufficient activity and selectivity in the presence of water and oxygen, respectively, when compared to the cobalt system.
The best catalyst to date is estimated to be lower in activity by a factor of four in wet stream [17]. Since any combustion process produces approximately 16% of water vapour, suggested catalyst should be stable in wet environment for a long time.
Zeolites containing non-transition metals, e.g. Ga– and In–exchanged ZSM5
and FER [27–29] have also been studied as CH4 – SCR catalysts. Although Ga-ZSM5
and Co-ZSM5 have comparable NO conversions, the temperature of maximum NO conversion is maintained by 100 K lower with Co-ZSM5 than with Ga-ZSM5. This temperature is reported as 673 – 723 K for the Co-ZSM5 catalyst [16,21,30]. In wet feed stream, the loss of catalytic activity is larger for Ga-ZSM5 than for Co–ZSM5 [21,28,30,31]. Although indium–exchanged ZSM5 promoted by precious metals is less sensitive to water vapour [32], the temperature of maximum NO conversion is too high for a practical application.
In conclusion, the zeolite based catalysts are sensitive to water, which makes them to deactivate quickly. Deactivation of the zeolite catalysts takes place by which temperature–induced ion migration and sintering of the active sites [33,34]. Another process leading to deactivation is the destruction of the zeolite structure, caused by
dealumination, that is, the removal of tetrahedrally coordinated Al3+ ions from the
zeolite lattice [35,36]. This process is accelerated by water. As a consequence of these all degradation processes, the chemisorption of the reactant molecules changes and inhibits the activity and selectivity of the catalyst. Long term hydrothermal testing [37] shows that the low hydrothermal stability of the zeolite catalysts is a serious
1.4 Selective Catalytic Reduction (SCR) of NOx over Non–zeolitic
Oxides with Methane
Limitations of the zeolite catalysts in the SCR of NOx promoted an extensive
search for non–zeolitic catalytic systems. Many metal oxides have been shown to be
active in the catalytic reduction of NOx with methane [10,38]. Only few of them
reduce NOx selectively in the presence of an excess of oxygen, which is the condition
for a practical application. Some of these systems are lanthanide oxides [39–42],
Group II A [40], Group IIIA [31,43], Group IIIB [44,45] metal oxides, SnO2 [46] and
supported transition metal oxides [47–54]. These catalysts possess lower activity in
the CH4–SCR than the zeolitic systems but they have better hydrothermal stability.
Palladium–based catalysts are well known for their ability to activate methane. Palladium is also an important constituent of the three-way catalysts (TWC)* for controlling automobile emissions. More strict emission regulations force most of the major automobile manufacturers to use pure palladium and promoted palladium catalytic converters in several parts of North America and Europe [48]. As a result of
the studies on this subject, it is reported that Pd/TiO2 catalyst shows nearly complete
conversion of NO with CH4 in the presence of oxygen. The activity of the catalyst is
not affected by water (up to 6%) [49-52] and higher selectivities were observed at higher water concentrations in the feed. However, the maximum conversion
temperature is too high (773 K) for actual NOx emission control [49]. Also formation
of ammonia, which is an undesired product, is observed [49].
It should be noted that cobalt supported on metal oxides, such as SiO2 and
Al2O3, prepared by impregnation with cobalt(II) nitrate solution are inactive in the
CH4–SCR [55,56]. On the other hand, Inaba et al. [53,54] have showed that the
preparation of the same catalysts by impregnation of the supports with a cobalt(II)
acetate solution results in materials exhibiting significant activity in the CH4–SCR
under anhydrous conditions. Application of cobalt(II) acetate solution ensures high dispersion of the cobalt ions. It is concluded that the presence of highly dispersed
cobalt ions is necessary for the CH4–SCR [53,54]. Utilization of a nitrate precursor
facilitates the agglomeration of cobalt species and leads to formation of unselective
Co3O4–like particles [53,57]. The latter are highly active for the combustion of
*TWCs are not selective toward NOx in the presence of oxygen. The reducer,
i.e. the hydrocarbon, is burnt with oxygen, and then the produced COy species react
with NOx species.
1.5 Mechanism of the SCR of NO with Methane in Excess Oxygen
Studies on the mechanism of NOx reduction with methane are mainly devoted to
the Co–zeolite systems. In 1995, Cowan et al. [58] reported that the rate ratio, CH4
consumption versus CD4 consumption, in the SCR of NO in the presence of an excess
oxygen over Co–ZSM5 catalyst is 2.40 at 648 K and 2.03 at 704 K by using a dry
feed and in the presence of 1.6% H2O, respectively. The expected values for a
primary kinetic isotope effect, calculated on the basis of the zero point energy
difference between the symmetric stretching frequencies of CH4 (2917 cm–1) and CD4
(2109 cm–1) are 2.44 at 648 K and 2.27 at 704 K. These values are close to the
experimental ones for the reaction in the absence and presence of water at those temperatures. As a conclusion, the rate-determining step of SCR of NO in the
presence of excess oxygen is the breakage of C – H bond by adsorbed NO2 to form a
methyl species. The following step is suggested by Cowan et al. [58] and Li et al. [18]
NO2 , ads + CH4 CH3• + HNO2 , ads (2)
which occurs on Lewis zeolite – Co2+ sites. Then the methyl radicals can react with
the chemisorbed NO2 to form the intermediate, nitromethane [58]. It is suggested that
the formed nitromethane further reacts toward N2 [59]. Studies of Liese et al. [60] on
the CeO2–H–ZSM5 system showed that a short–lived intermediate, such as nitro or
nitrosomethane is formed by interaction of adsorbed NO2 or nitrate with methane on
the CeO2 surface. This intermediate is detached from the CeO2 sites and transformed
to dinitrogen and carbon oxides over the BrØnsted sites of the ZSM5–based catalyst.
Yan et al. [61] proposed that the NO2 is generated on Co2+ ions and then, the
reduction with CH4 takes place over the BrØnsted sites of the zeolite. This conclusion
is based on the results of the CH4–SCR of NO study performed on a physical mixture
of Co/Al2O3 and H–zeolites.
Lukyanov et al. [62] concluded that the role of O2 is to convert NO to NO2. The
CH4–SCR on zeolite based catalysts in the presence of oxygen can be considered as a
In conclusion, the high performance of the zeolitic catalysts is associated with the peculiarity in the coordination of the exchanged metal ions, that act as strong Lewis acid sites, and the existence of considerably strong Brønsted acidity [10].
Only few papers are published dealing with the mechanism of CH4–SCR on
non-zeolitic catalysts [44,30,63,64]. As in the case of the zeolites the formation of the methyl radicals is the most important step in the process. Methyl radicals react with
the adsorbed NOx species to produce HCN. Further reactions of HCN produce CN
and NCO. These two intermediate species transform to N2, CO, CO2 and H2O. Group
III B metal oxides and lanthanide oxides are effective catalysts for the oxidative coupling of methane [65]. So, it is expected that methyl radical species are formed under SCR conditions as well.
Xie et al. [66,67] examined the SCR of NOx with methane on the Sr/La2O3 and
Ba–doped MgO catalysts. They reported significant activities for these catalysts. Over both catalysts, the formation of gas phase methyl radicals that react with NO in the gas phase to form nitrosomethane is proposed.
1.6 Non–zeolitic Solid Acid Supports for CH4–SCR Catalysts
Formation of organic radicals has been observed during isomerization of light alkenes catalyzed by sulfated [68,69] zirconia. This material possesses strong Lewis and Brønsted acidity [70] and according to some researchers [70,71] it is classified as superacid by using the Hammet indicator technique. Surprisingly, only a few reports
have been published regarding the activation of methane in the SCR of NOx in the
presence of supported oxides on non-zeolitic solid acids such as sulfated zirconia [43,72–77]. Most of the catalysts studied contained supported noble metals [72–
75,77]. The performance of gallium- and cobalt-supported on ZrO2-SO42− has been
investigated by Feely et al. [43] and Ciambelly et al. [76], respectively. In general, the
SO42−-modified catalysts are more active than the unmodified materials. The
performance of the catalysts varies much depending on the loading and dispersion of the active metal. It has been concluded that the acidity of the support is more
important in determining the activity of the catalyst than the absence of zeolitic structure [74]. The sulfate-modified catalysts possess good hydrothermal stability [73,75,77]. However, the maximum activity has been achieved in the 723 – 873 K
temperature range and data about N2O formation have not been reported, although it
is well known that supported noble metals when used as catalysts in the SCR of NO usually produce considerable amount of this greenhouse gas [10].
1.7 Acidity and Reactivity of Modified Zirconias
Hino and Arata [71,78] were the first to report that ZrO2 modified with sulfate
ions develops strong acidic properties and unique acid catalytic activity. Hammett
acid strength of sulfated zirconia is H0 = -16.04, whereas for 100% sulfuric acid this
value is only -11.99 [70]. Therefore, this material has been classified as solid superacid. The use of Hammet indicators is considered unreliable by most investigators for accurate measurement of solid acidity [79–85]. Babou et al. [83] and Kustov et al. [84] examined the sulfate groups on sulfated zirconia by IR spectroscopy and concluded that their acidity is not higher than the acidity of sulfuric acid or H-zeolites. Similar results are obtained by Drago and Kob [85]. Spectroscopic studies of Brønsted and Lewis acid sites of sulfated zirconia by means of solid-state NMR [86] and FTIR spectroscopy of CO adsorbed at low temperatures [87–90] showed that the Brønsted acidity of this material is clearly less than that of H-ZSM5, the latter being only moderately strong acid [91]. Furthermore, the Lewis acid sites on the modified zirconias are not superacidic and the strength of the Lewis acid sites on sulfated and
phosphated ZrO2 is similar [88], but stronger than those on tungstated zirconia
[87,89,90,92]. Thus, these systems are regarded as moderately strong solid acids. Promotion of sulfated zirconia with iron, manganese or platinum results in acidity strength that does not exceed that of the unpromoted materials [79,85]. On the other hand, the promoted catalysts exhibit higher activity and selectivity in the alkane isomerization than the unpromoted materials [81,93-97]. This fact suggests that the initiation of the reaction is different from the Olah-type chemistry in acidic solution, i.e. protonation of alkane. Recent investigations [69] show that the isomerization reaction of light alkanes on sulfated zirconia is initiated by homolytic cleavage of the R−H bond:
followed by one-electron transfer from the organic radical leading to formation of
carbenium and Zr3+ ions. It is suggested that in this process strong acceptor sites are
involved.
Concerning the nature of the acid sites involved in the alkane isomerization, it is a general consensus that properly activated sulfated zirconia contains both Brønsted and Lewis acid sites [70]. Discrepancies can be found on the predominant role played by a particular site with respect to the final activity showed by this material.
Figure 1 illustrates the model proposed by Arata and Hino [98] based on IR data. The formation of Brønsted sites is due to residual amounts of water.
Figure1: Model proposed by Arata and Hino [98].
Kustov et al. [84,99] postulated that the sulfate treatment of hydrous zirconia results in elimination of terminal ZrOH species by bisulfate ions. This leads to increase in the acid strength of the bridging ZrOH groups (Fig.2). The Lewis acidity enhancement was attributed to the increase of the electron-accepting properties of
three-coordinate zirconium cations via inductive effect of SO42– ions, which withdraw
Figure 2: Model proposed by Kustov et al. [84,99].
Figure 3: Model proposed by Adeeva et al. [100].
The model proposed by Adeeva et al. [100] is shown in Fig. 3, which represents sulfated zirconia acting as Brønsted and Lewis acid. According to these authors, the acid strength of the Brønsted sites of this material is similar to that of the lower OH-frequency protons in HY, but is weaker than that of the protons in H-ZSM5.
The final catalytic properties of sulfated zirconia in the isomerization of alkanes depend strongly on the preparation conditions. This material usually is prepared by a two step process, which involves mixing of zirconium hydroxide with sulfuric acid or
ammonium sulfate and then calcining the mixture to temperatures of 773–873 K [70]. It appears that one of the key points in preparing active catalysts by a two-step procedure is the amount of tetragonal phase in the sulfated zirconia. The catalytic activity is higher when the tetragonal phase is larger [70]. The tetragonal phase is stabilized by the sulfate groups on the surface, and is responsible for the increase in the surface area of the sulfated material.
1.8. Concluding Remarks
In the previous section it has been shown that the activation of light alkanes in the isomerization process starts with cleavage of the C-H bond. This process plays a crucial role in the activation of methane in the SCR of NO (section 1.5). It can be concluded that the catalysts that are active in the isomerization of saturated
hydrocarbons with lower carbon number could be active in the CH4-SCR of NO.
Results supporting this idea are summarized in section (section 1.6). However, further studies are needed in order to develop a durable catalyst that possesses high activity and selectivity at low temperatures for application under actual exhaust conditions.
From the kinetic studies on the CH4-SCR of NO over Co-ZSM5 catalyst [22,62]
it is known that the reaction order is higher toward CH4 than toward NO:
rNO = kPNO0.45 PCH40.62
This suggests that CH4 reacts with NO pre-covered catalyst. Generally the
adsorbed NO and NO2 (produced by oxidation of NO with O2) are weakly bonded to
the catalyst surface and desorb easily. Meunier et al. [101] showed that Co (of low loading) supported on alumina is not significantly active for the gas-phase oxidation
of NO to NO2 at the temperatures, at which this material is active and selective for the
SCR of NO with propene. Catalysts, which show good activity for NO oxidation, are
unselective for the reduction of NO in an excess of oxygen. The high rate of NO2 (g)
formation is related to high activity of the catalysts for hydrocarbon combustion [101]. These facts suggest that the key to the high selectivity and activity of the
catalysts for SCR of NO could be attributed to the reactivity of adsorbed NOx species.
The stable species formed after NO+O2 co-adsorption are the NOx– (x = 2, 3)
compounds [102]. It has been established that the latter readily interact with hydrocarbon reducers [101,103–108]. Moreover, different species possess different reactivities. For example, surface nitrates formed on Cu-ZSM5 are inert toward
interaction with methane, whereas CH4 is oxidized by Co-NO2– species formed on the
Co-ZSM5 catalyst [104]. This accounts for the lack of activity of copper-containing
samples in the CH4-SCR and the high activity of cobalt-based catalysts in this
reaction. These facts illustrate that the identification of the CH4-SCR reaction
intermediates and determination of the way of their formation and transformation to reaction products are critical in achieving selective performance of the catalysts. To elucidate these key issues, it is preferable that the reaction intermediates are stable enough to be detected spectroscopically.
IR spectroscopy is one of the most powerful tools for in situ characterization of
adsorbed NOx species on catalytic materials. Although the structural characteristics of
sulfated zirconia are well-documented [70,79], a few papers deal with IR
spectroscopic characterization of NOx species adsorbed on this material [109,110].
The results of recent investigations [110] have shown that the nitrate species formed
on zirconia during NO+O2 adsorption possess high thermal stability, which decreases
after introduction of sulfate ions and ions of transition metals. Another important observation is that the inherent Lewis acidity of the active sites in the modified
zirconia is additionally enhanced by the strongly adsorbed surface NOx species [110].
This can facilitate the activation of methane.
The aim of this study is to investigate, by means of in situ FTIR spectroscopy,
the reactivity of NOx species preadsorbed on catalysts containing cobalt(II) supported
on zirconia and sulfated zirconia toward methane. From these results the potentials of
these oxide materials as nonzeolitic catalysts for CH4-SCR of NO will be evaluated.
1.9 Identification of Strongly Adsorbed NOx– Species (x=2 or 3) by
FTIR Spectroscopy
The identification of adsorbed NOx species on the surface of the catalysts for
SCR of nitrogen oxides is a difficult task. Very often, the adsorption of NO and its co–adsorption with oxygen on oxide surfaces produce large number of adsorbed species exhibiting absorption bands that overlap. Table 1 summarizes the reported
frequencies of strongly adsorbed NOx species more frequently observed on metal
oxides and their corresponding structure [41].
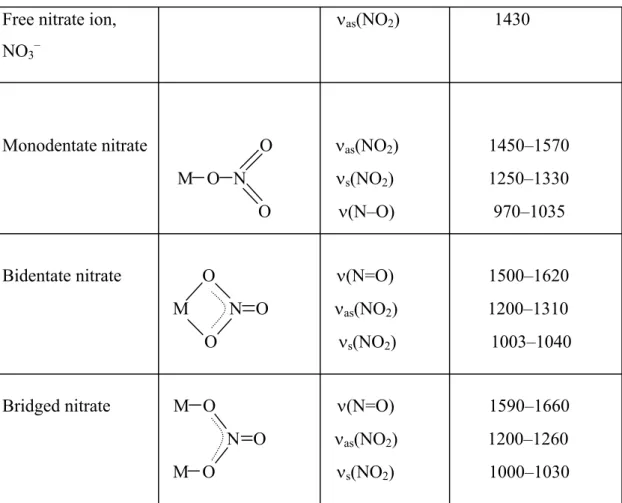
The free nitrate ion is planar with D3h symmetry and exhibits three IR active
bending (ν4) at 722 cm-1 [111]. Only the νas(NO2) mode lies above 1000 cm-1 and
could be detected on oxide surfaces. The νs(NO2) vibration is Raman active and is
observed at 1050 cm-1. After coordination, the symmetry of the NO3– ion is lowered to
C2v or Cs. As a result, the νs(NO2) mode becomes IR active and is observed around
1050 – 1000 cm-1. The degeneracy of the νas(NO2) vibration is removed and it splits
into two components. The magnitude of the split depends on the type of coordination and is larger for bridged and bidentate nitrates than for monodentate species (Table 1).
The free nitrite ion, NO2–, is characterized by νas(NO2) and νs(NO2) at 1335 and
1250 cm-1, respectively [111]. However, there are various possibilities for bonding of
NO2– species to the surface [41,102]. When coordinated via one or two oxygen atoms,
the NO2– ion forms nitrito species (Table 1). When the NO2– ion is bound through its
N atom, the nitro structure results. The data in Table 1 show that the differentiation between nitro-nitrito complexes and monodentate nitrates is difficult because of
overlapping absorption bands in the 1350 – 1550 cm-1 region, especially when the
sample is opaque below 1300 cm-1. In addition, the position of the IR bands does not
allow exact identification of the bridged and bidentate nitrates. As a result, different interpretations are offered in the literature and there is no agreement about the nature
of the adsorbed NOx species when dealing with a given oxide system.
Important information on the nature of the NOx species can be obtained from
their stability [102]. Usually, molecules such as NO, NO2, N2O3 are weakly adsorbed
and are easily removed by evacuation at room temperature. The ionic species (NO3–,
NO2–, NO+) display higher stability and require more energetic conditions to be
desorbed. In addition, the nitro species are more stable than the nitrito species and monodentate nitrates. Among the surface nitrates, the bridged and bidentate nitrates
possess higher stability than the monodentate NO3– species.
Analysis of the combination bands of the nitrate species shows that this spectral region can be used for the structural identification of bidentate and bridged nitrates
[110]. Bridged nitrates produce combination bands at 2845 – 2800 cm-1 [ν(N=O) +
νas(NO)2] and pair of bands at 1980 – 1960 cm-1 [νs(NO2) + δ(ONO)]. Bidentate
nitrates can be distinguished by the appearance of combination bands in the region of
2600 cm-1 [ν(N=O) + νs(NO2)] and pair of bands between 1755 and 1700 cm-1
Table 1: Frequencies of Adsorbed NOx Species Observed on Metal Oxides and Their
Corresponding Structure [41].
NOx Species Structure Vibration mode Wavenumber[cm-1]
Free nitrite ion, νas(NO2) 1250
NO2– νs(NO2) 1335 δ(ONO) 830 Nitro O νas(NO2) 1370–1470 M–N νs(NO2) 1320–1340 O δ(ONO) 830 Nitrito ν(N=O) 1400–1485 M–O O ν(NO) 1050–1100 N δ(ONO) 820–840
Chelated nitrito O νas(NO2) 1270–1390
M N νs(NO2) 1170–1225
O δ(ONO) 840–860
Bridging nitro M M νas(NO2) 1390–1520
N O νs(NO2) 1180–1260
O
Bidentate nitro O νas(NO2) 1390–1520
N O νs(NO2) 1180–1260
Table 1 continued
Free nitrate ion, νas(NO2) 1430
NO3–
Monodentate nitrate O νas(NO2) 1450–1570
M O N νs(NO2) 1250–1330
O ν(N–O) 970–1035 Bidentate nitrate O ν(N=O) 1500–1620
M N O νas(NO2) 1200–1310
O νs(NO2) 1003–1040
Bridged nitrate M O ν(N=O) 1590–1660
N O νas(NO2) 1200–1260
2. EXPERIMENTAL
2.1 Sample Preparation
Zirconia was prepared by hydrolysis of ZrCl4 (Merck, for synthesis) with a
concentrated (25%) solution of ammonia. The pH of the solution is reached to 10.5
due to ammonia addition. During the dissolution of ZrCl4 in water (0.3 mol of
ZrCl4/L) and addition of ammonia solution, the temperature was kept below 343 K.
The precipitate was washed thoroughly with doubly deionized water to remove the
chloride ions (AgNO3-tested), dried at 393 K for 12 h. This material represents
hydrated zirconia, Zr(OH)4.xH2O.
Sulfated zirconia was prepared by impregnation of the hydrated zirconia with a
solution of (NH4)2SO4. Two samples were prepared containing different amount of
sulfate ions, i.e. 4.5 and 20 wt %. These materials were dried at 373 K and calcined for 2h at 623 K and 2h at 773 K.
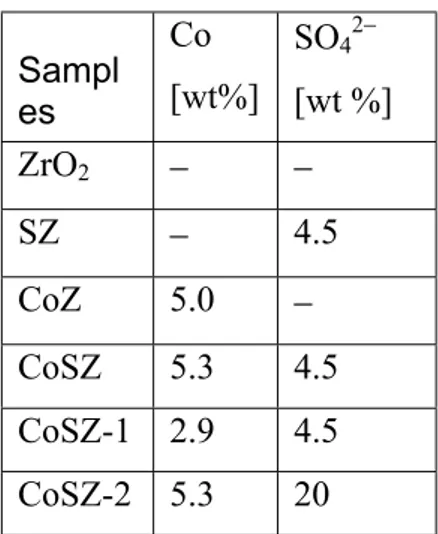
The cobalt-containing samples were prepared by wet impregnation of either hydrated or sulfated zirconias using cobalt(II) acetate as a precursor. The samples were dried and calcined according to the same procedure applied to the sulfated zirconias. The nominal content of the active components and the notation of the samples is given in Table 2.
Sampl es Co [wt%] SO42− [wt %] ZrO2 − − SZ − 4.5 CoZ 5.0 − CoSZ 5.3 4.5 CoSZ-1 2.9 4.5 CoSZ-2 5.3 20
2.2 Surface Area Measurements and X–ray Diffraction
The BET surface areas of the samples (dehydrated at 523 K) were measured by nitrogen adsorption at 77 K using a MONOSORP apparatus from Quanto Chrome.
XRD analysis was performed on a Rigaku Miniflex diffractometer with Ni–
filtered Cu Kα radiation under ambient conditions. The scan speed was 10/min for the
samples studied, except for the CoSZ. In the latter case the scan speed of 0.50/min was
used.
2.3 IR Spectroscopy
The FTIR spectra were recorded on a BOMEM MB 102 FTIR (Hartman & Braun) spectrometer equipped with a liquid–nitrogen–cooled MCT detector at a
resolution of 4 cm-1 (128 scans).
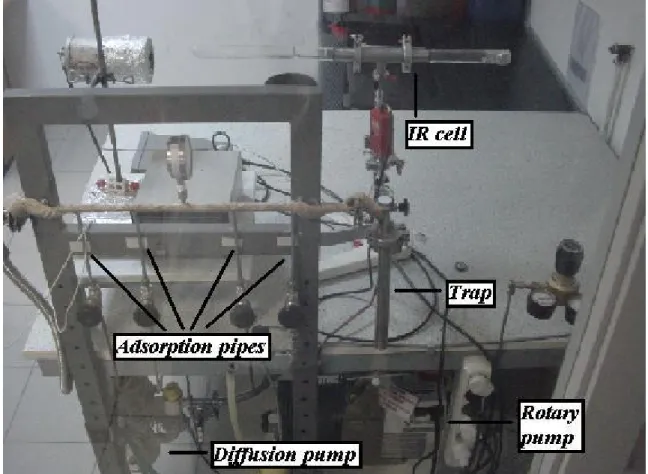
2.4 Experimental Setup
A specially designed IR cell allowed registration of the spectra at ambient temperature and catalyst activation at higher temperatures (Figure 4). The cell was connected to a vacuum/adsorption apparatus (Figure 5). It has a sample introduction part and end, which allows heating. At the other end of the cell, NaCl windows are installed, which allows recording of the IR spectra.
2.5 Activation of the Samples
Self–supporting discs (0.026–0.030g/cm2) were used for the FTIR studies.
These specimens were activated by heating for 1 h in a vacuum at 673 K and in oxygen (13.3 kPa) at the same temperature, followed by evacuation for 1 h at room temperature.
The spectra of the activated samples (taken at ambient temperature) were used as a background reference. The spectra of the samples that were subject to treatment at elevated temperatures were recorded after the IR cell had been cooled to room
temperature. All of the spectra presented (except those in Figure 7, Section 3.1.2) were obtained by subtraction of the corresponding background reference.
Figure 4: IR cell and the furnace.
2.6 Adsorption of NO and Co–adsorption with O2
NO adsorption was performed at room temperature by introducing 0.67 kPa of NO and the evolution of the IR spectra with time was followed.
Co–adsorption with O2 was achieved at room temperature by introducing a gas
mixture (1.33 kPa) of NO and O2 (1:1) for a given period of time.
Thermal stability of the adsorbed NOx species was studied by heating the
sample for 15 min under vacuum in the temperature range 373–723 K.
The computer peak fittings were performed using the minimum number of peaks and fixed peak positions based on the original spectra.
2.7 Interaction of CH4 with Catalysts
Interaction of CH4 with catalyst was observed by addition of 6.7 kPa of CH4
into the IR cell. Then the closed IR cell was heated gradually from 453 K to 723 K. At each temperature level the interaction time was 30 min.
2.8 Interaction of CH4 with NOx pre–covered catalyst
After the formation of the NOx via co–adsorption of NO and O2 (1:1) followed
by the evacuation of the gas phase for 5 min, 6.7 kPa of CH4 was introduced to the IR
cell. The interaction of the NOx species with methane was studied in the temperature
range of 453 – 723 K. The interaction time at each temperature level was 30 min.
CH4 and O2 used were passed through a trap cooled by liquid nitrogen before
3. RESULTS AND DISCUSSION 3.1 Characterization of the Samples
3.1.1 BET Surface Area Measurements and X-ray Diffraction
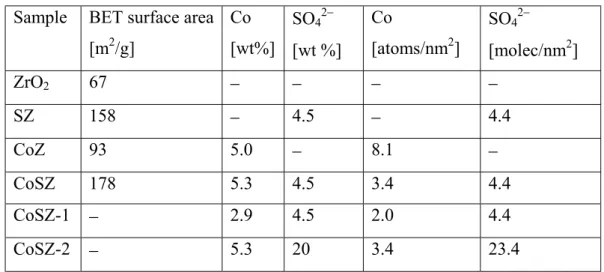
In Table 3 the content (in wt %) and the surface concentrations of the active
components (number of molecules/nm2) are given together with the surface areas of
the samples studied and supports used.
Table 3: Surface areas and loading of the sample studied*
Sample BET surface area
[m2/g] Co [wt%] SO42− [wt %] Co [atoms/nm2] SO42− [molec/nm2] ZrO2 67 − − − − SZ 158 − 4.5 − 4.4 CoZ 93 5.0 − 8.1 − CoSZ 178 5.3 4.5 3.4 4.4 CoSZ-1 − 2.9 4.5 2.0 4.4 CoSZ-2 − 5.3 20 3.4 23.4
*All materials are calcined for 2 h at 623 K and 2 h at 773 K.
For calculation of the surface concentration of the sulfate ions, the average
cross-section of an adsorbed sulfate ion equal to 0.25 nm2 [112] was used. The surface
concentration of the sulfate ions corresponding to the theoretical monolayer is
estimated to be 4.0 SO42–/nm2. The theoretical loading of cobalt on zirconia
corresponding to the monolayer (3.5 Co/nm2) was determined using the fact that there
are 7 Zr4+ ions/nm2 on complete dehydroxylated monoclinic zirconia [113] and
assuming two anchoring zirconium sites per one cobalt atom. According to Morterra et al. [114] at surface concentrations of the sulfate ions equal or larger than the
theoretical monolayer, all coordinative positions of the Zr4+ ions are saturated. This
means that the surface concentration of the cobalt ions on the CoSZ sample
corresponding to the theoretical monolayer should be equal to 4 Co atoms/nm2. The
into account the surface area of the zirconia and sulfated zirconia, respectively, calcined at 773 K. Surface concentration of the sulfate ions in the sulfated zirconia and CoSZ sample was estimated using the surface area of the calcined zirconia.
Preliminary experiments showed that among the Co/sulfated zirconia
materials, the reactivity of the NOx species adsorbed on the sample containing 5.3
wt% of cobalt and 4.5 wt % of sulfate ions (CoSZ) is the highest and this catalyst was
investigated in detail. The surface concentration of the sulfate ions (4.4 SO42−/nm2) in
the CoSZ sample practically corresponds to the theoretical monolayer, whereas the
coverage by cobalt ions (3.4 Co/nm2) is slightly lower than the theoretical monolayer.
In order to understand the effect of the sulfate ions on the performance of the cobalt(II)-zirconia system, the same amount of cobalt (5.0 wt%) was introduced into sulfate-free zirconia (CoZ catalyst).
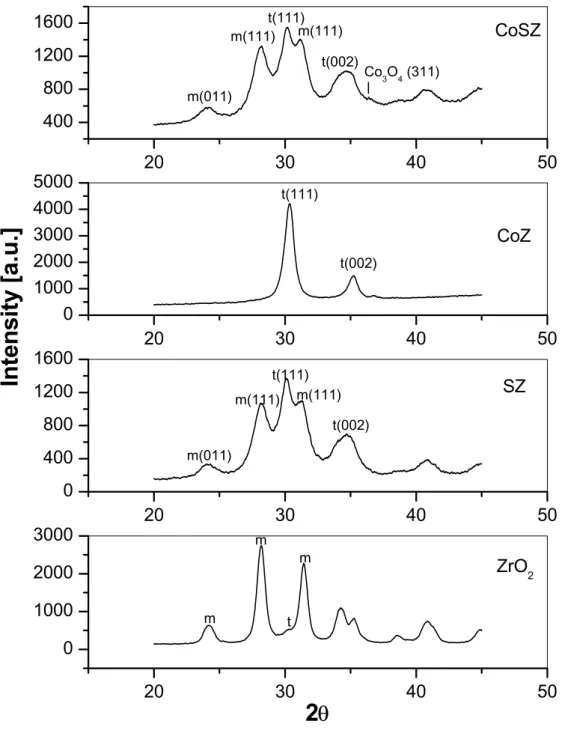
Figure 6 illustrates the powder X-ray diffraction (PXRD) patterns of the catalysts studied and supports used (calcined at 773 K). The crystallographic phase compositions were calculated by using the method of Toraya et al. [115]. According
to this method, the integrated areas under the resolved reflections (It, Im) were used to
calculate the intensity ratio, Xt (Eq. (1)). Then the tetragonal volume percentage, Vt, is
calculated by using an empirical non–linear relationship (Eq. (2)) obtained from
physical mixtures of monoclinic and tetragonal ZrO2.
It(111) (1)
Im(111) + Im(111) + It(111)
1.311 (1–Xt) (2)
1.311 – 0.311 Xt
According to these calculations, the tetragonal volume percentages are calculated as 9% and 67% for the zirconia and sulfated zirconia, respectively. Introduction of cobalt to the pure zirconia converts the structure of the support from predominant monoclinic to 100 % tetragonal, whereas the deposition of cobalt does not have effect on the phase composition of the sulfated zirconia. The CoSZ catalyst is 67 %
tetragonal. The XRD pattern of the CoZ catalyst contains weak peak at 2θ = 36.80,
which is attributed to the strongest (311) reflection of cubic Co3O4 [116]. In the case
of CoSZ sample, there is a very weak feature at approximately 2θ = 36.40. This
Xt =
Figure 6: Powder X–ray diffraction patterns of the ZrO2, SZ, CoZ, CoSZ samples.
(m: monoclinic, t: tetragonal)
reflection is not observed in the case of the SZ sample. Therefore, it is concluded that
small amount of Co3O4 is present on the surface of the CoSZ sample as well.
20 30 40 50 0 1000 2000 3000 t m m m ZrO2
Inte
n
sity
[a
.u
.]
2
θ
20 30 40 50 0 400 800 1200 1600 t(002) m(011) m(111) m(111) t(111) SZ 20 30 40 50 0 1000 2000 3000 4000 5000 t(002) t(111) CoZ 20 30 40 50 400 800 1200 1600 Co3O4 (311) t(002) m(111) t(111) m(111) m(011) CoSZThe broad diffraction patterns of the SZ and CoSZ samples can be attributed to the reduction of the grain size. The crystallite size was calculated using the Sherrer equation [117] (equation 3). In this method, the relationship between the band structure and crystallite size is used. Here, the interference of the reflected photons from different crystal planes is the important feature. Constructive interference (in phase) of the two reflected photons is maintained at a specific angle, which is called
Bragg`s angle, θB. Deviations from this angle results in the partially out of phase
contribution, which causes decrease in the intensity of the beam. If the size of the crystal increases, the number of the reflected waves that are partially out of phase increases. Thus the lower intensity of the reflected beam for angles slightly different from Bragg`s angle will be observed. So the full width at half-maximum (FWHM) value will decrease and sharper peaks will be observed for bigger crystals in the PXRD pattern.
S = 0.9λ / (B.cos θB) (3)
where the S is the size of the crystallite, B is the FWHM value and λ is the
wavelength of the source. The 2θB values are 30.4 and 30.30 for the CoZ and CoSZ
sample, respectively. The calculated crystallite sizes are 10.8 nm for the CoZ catalyst and 8.0 nm in the case of the CoSZ sample.
3.1.2 FTIR Spectra of the activated samples
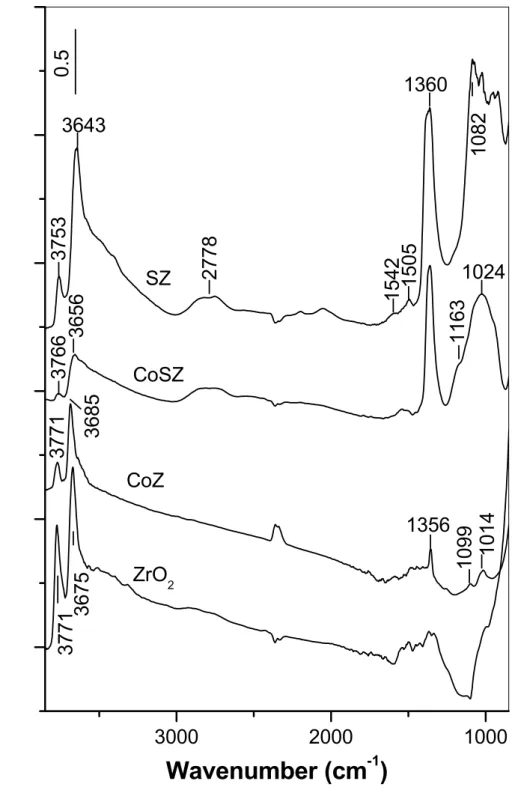
Figure 7 shows the FTIR spectra of the catalysts CoZ and CoSZ together with the spectrum of pure zirconia and sulfated zirconia (SZ). The spectra are normalized by a factor accounting for the difference between the surface area of zirconia and the
surface areas of the CoZ, SZ and CoSZ samples. The spectrum of the ZrO2 support in
the OH stretching region displays a pair of sharp bands at 3771 and 3675 cm-1.
According to the literature data [110,118,119] these bands are assigned to terminal
(3771 cm-1) and bridged (3675 cm-1) hydroxyls coordinated to three Zr atoms. The
broad band between 3600 and 3000 cm-1 is attributed to H-bonded hydroxyls.
The impregnation of zirconia with aqueous solution of cobalt(II) acetate (CoZ sample) results in decrease in the intensities of the bands of both terminal and bridged OH groups and decrease in the population of H-bonded hydroxyls. This accounts for their participation in the deposition process.
Figure 7: FTIR spectra of the activated catalysts and supports.
Modification of zirconia with sulfate ions (sample SZ) affects mainly the intensity of the band due to terminal hydroxyls. In the CoSZ sample, the additional
decrease in the intensities of the bands at 3771 and 3656 cm-1 is associated with
anchoring of the cobalt ions to both types of zirconia hydroxyls.
3000
2000
1000
3766
3771
2778
1082
3643
SZ
3771
ZrO
23675
0.
5
3685
1014
1099
1356
1024
1163
1360
1505
1542
3753
3656
CoZ
CoSZ
Absor
b
ance
Wavenumber (cm
-1)
The complex band at 1360 cm-1 with a shoulder at about 1400 cm-1 observed on the sulfated samples corresponds to the ν(S=O) vibration of highly covalent
sulfates coordinated to Zr4+ ions, whereas the absorption in the 1200 – 870 cm-1
region is assigned to ν(S−O) stretching vibrations [120]. The shoulder at 1163 cm-1
observed in the spectrum of the CoSZ sample reveals the presence of more ionic sulfates attached to cobalt sites. Occurrence of transformation of covalent sulfates to
more ionic structures is confirmed by the decrease in the intensity of the 1360 cm-1
band in the CoSZ catalyst relative to that in the SZ sample.
Sulfate-containing samples exhibit broad absorption between 3600 and 3000
cm-1 typical of H-bonded hydroxyls of the type O−H :O−H. In addition, another,
broad band between 3000 and 1900 cm-1 can be seen in the spectra. This absorption is
attributed to OH groups (most probably tribridged) involved in H-bonding with the
oxygen atoms of the surface sulfate groups. The distinct minimum at 2630 cm-1
divides the broad band into two pseudobands, the so-called A and B bands [121–123]. This minimum appears as a result of overlap between a sharp band due to the 2δ(OH) (in-plane bending) modes and the broad fundamental band corresponding to the perturbed OH groups. The missing intensity is redistributed to both sides of the minimum by Fermi resonance of the superimposed bands [121]. Similar bands (the so-called A, B, C triplet) have been observed upon absorption of basic molecules on zeolites [121–123]. In the case of the CoSZ and SZ samples, the C band is absent, which indicates that the interaction of the surface OH groups with the sulfate ions is medium-strong [123]. The A band for both samples has complex shape due to
superimposition of the 2ν(S=O) overtone band at approximately 2778 cm-1 to the
stretching vibrations of the perturbed OH groups. In the case of sulfated zirconia, the
first overtone of the ν(S−O) modes between 1090 and 1000 cm-1 causes analogous
complication in the shape of the B band (broad minimum in the 2180 – 2100 cm-1
region).
The residual OH bands in the SZ and CoSZ samples occur at lower frequency
than in the pure zirconia (3766 – 3753 cm-1 versus 3771 cm-1 and 3656 – 3643 cm-1
versus 3675 cm-1) indicating enhanced Brønsted acidity. In the CoZ catalyst only the
position of the bridged OH groups is affected. They are blue shifted compared to that on the pure zirconia indicating that the deposition of cobalt leads to reduced Brønsted
acidity. The CoSZ sample shows also lower Brønsted acidity relative to that of the sulfated zircona.
The bands between 1550 - 1400 cm-1, detected with variable intensities for the
zirconia and CoSZ and SZ samples, are assigned to split ν3 modes of surface
carbonates [124]. Such residual species are often observed on zirconia surfaces and cannot be removed by high-temperature activation of the catalyst. The sharp band at
1356 cm-1 and the weaker absorptions at 1099 and 1014 cm-1 observed on the CoZ
sample probably are due to overtones and combination bands of Co3O4 [125] in
agreement with the PXRD data.
3.2 NO and NO/O2 Co–adsorption and Thermal Stability of NOx
Species
3.2.1 Adsorption of NO at room temperature on CoZ sample
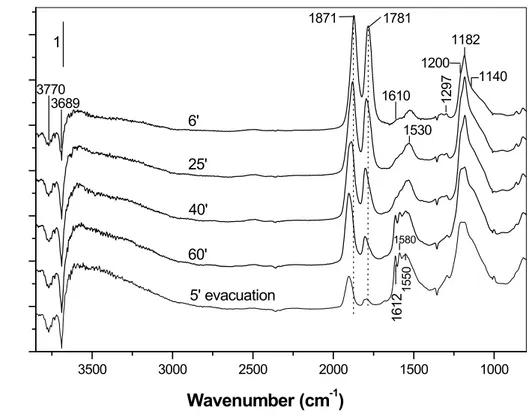
Evolution of the spectra of adsorbed NO (1.07 kPa) at room temperature on the activated CoZ sample with time is shown in Figure 8. The spectrum taken after 6 min of the introduction of NO to the IR cell, contains two strong bands in the nitrosyl
region positioned at 1871 and 1781 cm-1. Since the Zr4+−NO species are characterized
by absorption at 1912 cm-1 [110,126,127], the former two bands are associated with
the cobalt sites. According to the literature data [56,128,129], this pair of bands is due
to symmetric and asymmetric vibrations of dinitrosyls formed on Co2+ ion, i.e.
Co2+(NO)2 species. The bands at 1871 and 1781 cm-1 decrease in intensity and shift to
higher frequency. After 60 min they are positioned at 1902 and 1802 cm-1,
respectively. In the low-frequency region, another strong and complex band at 1182
cm-1 develops with time reaching maximum intensity after 25-40 min from the
beginning of the adsorption. According to the literature data [110], the absorption
with maximum at 1182 cm-1 corresponds to the ν(NO) stretching mode of anionic
nitrosyl, NO−, coordinated to Zr4+ sites. The NO− species are produced by
disproportionation of NO with the participation of the surface hydroxyls according to the reaction [110]:
2NO + 2OH− → NO− + NO
Figure 8: FTIR spectra of adsorbed NO (1.07 kPa) on the CoZ catalyst at room
temperature for various times. The spectrum of the activated sample is used as a background reference.
Involvement of the surface OH groups in the interaction with NO is supported by the
appearance of negative bands at 3770 and 3689 cm-1 and positive absorption in the
3660 – 3000 cm-1 region due to H-bonded OH groups. The band at 1610 –1612 cm-1
(not resolved at the beginning of the adsorption process) is assigned to the δ(HOH) modes of adsorbed water molecules. The subtraction spectra (not included) show that
the bands at 1200 and 1140 cm-1 behave synchronously and they are ascribed to the
νs(NO) and νas(NO) stretching vibrations, respectively, of the hyponitrtite ion, N2O22–,
[41]. The band at 1297 cm-1 is attributed to the ν(N=N) mode of N2O22– ion. This
absorption is usually very weak [41]. Keeping the sample in contact with NO for
longer time (60 min), results in decrease in the intensity of the band at 1182 cm-1
characteristic of the NO– species and increase in the intensities of the bands due to
N2O22– ion. This indicates that the hyponitrite ions are formed by dimerization of the
NO– species. 3500 3000 2500 2000 1500 1000 1550 1580 16 12 12 97 1530 1610 3689 3770 1140 1200 1182 1871 1781 1 5' evacuation 60' 40' 25' 6' A b so rb an ce Wavenumber (cm-1)
The bands at 1580 – 1530 cm-1 that develop with time of NO exposure are
attributed to the νas(NO2) of chelated NO2− (nitro) species [41]. The corresponding
νs(NO2) stretching vibrations are superimposed to the bands due to the NO− and
N2O22– ions and are not resolved. The nitro compounds are produced by both
disproportionation of NO (reaction 3) and oxidation of the adsorbed NO. The occurrence of the latter process is confirmed by the progressive decrease in the
intensities of the bands at 1871 and 1781 cm-1. It can be proposed that the adsorbed
NO is oxidized to bridged nitro species by cobalt(II) sites located in the vicinity of the cobalt(II) dinitrosyls (Scheme 1):
Scheme 1
The spectra in Fig. 8 show that the process of oxidation is slower than the disproportionation of NO.
Evacuation at room temperature for 5 min affects mostly the intensities of the
Co2+(NO)2 bands (Figure 8).
Careful inspection of the shape of the band at 1871 cm-1 (Figure 9) shows that
a high-frequency shoulder starts to develop after 25 min of the NO adsorption. Results of curve fitting of the spectra taken in the period from 25 to 60 min show that the
intensities of the bands at 1880 – 1887 and 1796 – 1803 cm-1 due to the Co2+(NO)2
species decrease with simultaneous growth of a band at 1913 – 1909 cm-1. The latter
band develops simultaneously with the absorption in the 1580 – 1400 cm-1 region due
to the nitro species. For this reason, the band at 1913 – 1909 cm-1 is assigned to the
ν(NO) stretching vibration in a complex of the type ON−Co2+−NO
2− (Scheme 1). The
NO2− species possess electron-accepting ability. As a result the electrophilicity of the
Co2+ ions increases. This leads to enhancement of the σ component of the bond
between the NO and the cobalt(II) site and stronger N−O bond in the ON−Co2+−NO
2− N ON O O Co+ O Co2+ O O O NO ON Co2+ O Co2+ O 2-O
Figure 9: Results of curve-fitting procedure applied to the FTIR spectra taken after
NO adsorption (1.07kPa) on CoZ sample at room temperature. The spectrum of the activated sample is used as a background reference.
complex. In other words, the ν(NO) stretching mode is at higher wavenumber than that expected for a simple cobalt(II) mononitrosyl (for example, in the case of
Co2+/SiO2 the ν(NO) stretching vibration is observed at 1865 cm-1 [102]). Based on
these observations, it can be concluded that the increasing time of NO exposure
causes transformation of Co2+ dinitrosyls to ON−Co2+−NO2− species. This conclusion
is supported by the observed linear correlation between the integrated absorbances of
the bands at 1909 – 1913 and 1880 – 1887 cm-1 corresponding to the ν(NO) and
νs(NO) modes of the ON−Co2+−NO2− and Co2+(NO)2 species, respectively (Figure
10). 2050 2000 1950 1900 1850 1800 1750 1700 1650 2050 2000 1950 1900 1850 1800 1750 1700 1650 2050 2000 1950 1900 1850 1800 1750 1700 1650 2050 2000 1950 1900 1850 1800 1750 1700 1650 2050 2000 1950 1900 1850 1800 1750 1700 1650 0.1 1796 1880 1913 25 min. 0.1 1796 1910 1881 30 min. 0.1 1801 1911 1886 40 min. 0.1 1803 1887 1910 50 min. Abs o rb ance Wavenumber (cm-1) 0.1 1803 1887 1909 60 min.
Figure 10: Correlation between the integrated absorbances of the bands at 1909-1913
cm-1 and 1880-1887 cm-1.
3.2.2 Coadsorption of NO and O2 on the CoZ Sample and Thermal
Stability of the NOx Species Produced
SCR reaction occurs under excess of oxygen. In the absence of oxygen the catalysts are either inactive or posses a low activity [11,130]. So, to understand the mechanism of SCR, it is necessary to study the adsorption of NO in the presence of oxygen.
Figure 11 shows the spectra of the CoZ sample obtained upon adsorption of
0.67kPa of NO for 5 min (spectrum a), followed by addition of 0.67kPa of O2 to the
IR cell for 30 min (spectrum b). Immediately after the introduction of the oxygen the color of the gas phase changed to brown. This is an indication that considerable
amount of NO2 was formed. This causes drastic changes in the spectrum of
preadsorped NO. The bands due to Co2+(NO)2 species disappear and absorption at
15 20 25 30 35 40 45 50 12 14 16 18 20 22 24 26 28 30 60' 50' 40' 30' 25' A 1880 -1 887 [a.u.] A1909-1913 [a.u.]
![Figure 1 illustrates the model proposed by Arata and Hino [98] based on IR data. The formation of Brønsted sites is due to residual amounts of water.](https://thumb-eu.123doks.com/thumbv2/9libnet/5788568.117702/26.892.172.683.362.585/figure-illustrates-proposed-arata-formation-brønsted-residual-amounts.webp)
![Figure 2: Model proposed by Kustov et al. [84,99].](https://thumb-eu.123doks.com/thumbv2/9libnet/5788568.117702/27.892.261.671.72.392/figure-model-proposed-kustov-et-al.webp)
![Table 1: Frequencies of Adsorbed NO x Species Observed on Metal Oxides and Their Corresponding Structure [41].](https://thumb-eu.123doks.com/thumbv2/9libnet/5788568.117702/31.892.130.758.119.1080/table-frequencies-adsorbed-species-observed-oxides-corresponding-structure.webp)