CİNSİYET FARKLANMASI ve
CİNSİYET KROMOZOMLARINA BAĞLI OLARAK
GELİŞEN GENETİK HASTALIKLAR
Cinsiyet gelişimini belirleyen faktörler;
1.Kromozomal durum (46, XX veya 46, XY) 2. Gonadal gelişim (over veya testis varlığı) 3. Fenotipik görünüm (dişi-erkek, internal ve
eksternal genitalya) 4. Hormonal düzey (östrojen-testesteron) 5. Psikolojik durum
Cinsiyetin erkek ve kadın olarak saptanabilmesi, bu etmenlerin hepsinin aynı yönde gelişmesi ile mümkündür.
Üreme (reprodüktif) Sisteminin Embriyolojisi
Erken embriyonik dönemde gonadlar 6. haftaya
kadar cinsiyet ayrımı göstermezler ve germ hücresi ihtiva etmezler.
Gonadal gelişim ya testis ya da over yönünde olur. Bu gelişimin tipi Y kromozomunun varlığına bağlıdır. Eğer bir Y kromozomu varsa erkek gonadı gelişir,
Gebeliğin 6. Haftasında primordial germ hücreleri ekstraembriyonik yerlerinden gonadal çıkıntıya göç ederler ve bir çift gonad oluşumu için seks kordları ile çevrilirler.
Bipotansiyel farklanmamış gonadlar medulla ve korteks bölümlerinden oluşur
korteks
over
medulla
Müller kanalı
(Paramezonefrik)
Kadın iç genital sistemi
Wolf kanalı (Mezonefrik)
Erkek iç genital sistem
Fallop tüpleri, Uterus, 1/3 üst Vajen
Vaz defferens,
seminal veziküller epididimis
Dış genital organlar da ortak dokuların
farklanmasıyla oluşur.
Genital çıkıntı
klitoris
Genital kıvrımlar labia minörler
Genital kabartı labia majörler
penis
penis gövdesi
Overyan gelisim, Y kromozomunun k
ı
sa kolundaki testis determinasyon faktörü (TDF) geni SRYbulunmadığı sürece oluşur.
Bu gen gelişimin erkek yönünde devam etmesinden sorumlu bir transkripsiyon faktörüdür.
TDF varsa;
Anti mülleryan hormon (MKİ) (gen 19. kromozomda yerleşik)
salınarak Mülleryan kanal inhibe edilir. Testosteron salınarak
dış genitalya maskülinizasyonu sağlanır.
Testosteron:
Fetal testisin intertisiyel Leydig hücrelerinden salgılanır.
Maskülinizan etkiyi 5 redüktaz enzimi ile dihidrotestesteron’ a çevrilirse gösterir.
İkisi birlikte androjen reseptörlerini etkileyerek; Dış genitalyanın genital çıkıntıdan penis ve testis yönünde değişimi,
iç genitalyada ise urogenital kanalların vas deferens yönünde değişimini sağlar.
Erkek yönünde genital gelişim için olması
gerekenler:
SRY taşıyan normal bir Y kromozomu
Testesteron sentezi
5 redüktaz enzimi ile dihidrotestesteron
sentezi
Androjen reseptörlerinin varlığı ve
fonksiyonel olması
Cinsiyet kromozomları belirleyici olmakla beraber,
bunların otozomal kromozomlardaki genlerle iletişim ve etkileşim içinde olması da önemlidir.
Cinsiyeti belirleyen kromozomlar ‘X’ ve ‘Y’
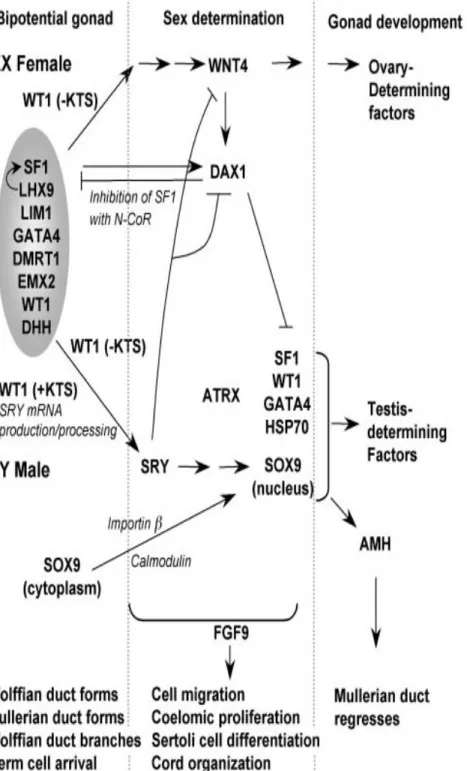
SRY geni düzenleyicileri;
SOX9 (17. kromozom)
DAX1 (Xq21)(anti SRY etki) SF1 (10q)
WT1(11p13) DMRT1(9q24)
DAZ like gen ailesi(3. kromozom) AMH(MIS) (19. kromozom)
WNT4(1p)
ATRX del10q del2p
FIG. 10. Model for initiation of sex determination. The positions of a number of genes believed to mediate key events in sex determination and differentiation are indicated and discussed in the text (Section VI).
WT1 geninin farklı mutasyonları cinsiyet farklanma
bozukluğu ile birlikte giden üç tip sendroma yol açar
Fraiser S
Denys-Drash S
WAGR (Wilms’ tumor, aniridia,
genitourinary abnormalities, and mental retardation) Kromozomal olarak erkek, Denys-Drash sendromlu hastalar dişi ya da belirsiz genitalyaya sahip olabilir. WT1 geni ürünü sertoli ve ledig hücreleri arasındaki ilişkiyi düzenler.
11p13 deki WT1 geninin dominant mutasyonları normal testis gelişimini engeller.
9p24 de yerleşik DMRT1 geninin de gonad gelişimi için önemi vardır.
9p delesyonu olan 46,XY bireylerde dişi fenotip veya belirsiz dış genitalya olabilir.
Trizomi 9p Klinefelter Sendromu benzeri fenotipe yol açabilir.
CİNSİYET KROMOZOM BOZUKLUKLARI VE
CİNSİYET FARKLANMASINI BOZAN
Sayısal (anöploidiler)
Yapısal (delesyon,translokasyon, izokromozom) Mozaiklik sık rastlanan bir durumdur.
500 doğumda 1 görülürler.
X inaktivasyonu ve Y kromozomunda yaşamsal önemi olmayan genlerin varlığı bu hastalıkların klinik olarak hafif olmasına neden olur
Trizomiler canlı doğumda en sık görülen bozukluklardır (XXX,XXY,XYY).
Monozomi X en sık spontan abortuslarda bulunur.
Cinsiyet farklanma kusurlarının %20 sinden kromozom bozuklukları sorumludur.
Y kromozomu
Heterokromatin Ykromatin Yp11.2 AZFa,b,c,d USP9 H-Y antijeniSRY geni mutasyonları
XY karyotipe sahip dişi bireyler saptandığında akla
gelmelidir.
Bu fenotipe sahip bireyleri %15 inde belirlenir, bu
nedenle SRY dışı erkek tipinde farklanmadan sorumlu
genler olduğunu destekler.
Yq azospermi ve spermatogenez ile ilgili genleri taşımaktadır.
Nonobstruktif azospermide %10, oligospermide daha az oranda Yq intertisiyel delesyon saptanmaktadır.
Bu delesyonlar de novo dur, kişinin babasında ve kardeşlerinde bulunmaz.
AZF delesyonları DNA elde edilerek moleküler olarak saptanabilir.
Toplumdaki genel sıklık bilinmemektedir.
Sperm üretiminde bozukluk olan infertil kişilerde bulunma riski %2 dir.
AZF a,b,c ve d olarak dört ayrı gruba ayrılır. Delesyonlar yama şeklinde bölgelerin bazılarını veya tümünü içerebilir.
X ve Y kromozomunda krosing-over hataları 46,XX erkekler
46,XY kadınlar
Y kromozom yapısal bozuklukları
Yp delesyonları
SRY geni kaybı ile sonuçlanır.
Klinik boyu kısa olmayan bir Turner sendromu kliniğidir.
Yq delesyonu
Erkek fenotip oluşur.
Bazı olgularda infertilite ile seyreder.
Azospermik erkeklerde %10-15 oranında bildirilmiştir.
İdic(Yp)/idic(Yq)
Genellikle 45,X bir hücre dizisiyle birlikte mozaik olarak saptanırlar
Sıklıkla Miks gonadal disgeneziye neden olurlar ancak fenotip değişkendir.
45,XO/46,XY Mosaisizm
(Miks gonadal disgenesis)
Bebek doğduğunda dişi, erkek tipinde ya da
ambigous (belirsiz) dış genitalyaya sahip olabilir.
Erkek tipinde olanlar normal gelişim sürdürürlerken
Dişi tipindekiler genellikle Turner sendromu fenotipi
oluştururlar.
Bunlarda gonadal tümör sıklığında artış olduğundan
X kromozomu
Kadınlarda iki tanedir,ancak tek X raslantısal olarak inaktive edilir
(Lyon hipotezi)
Döllenmeden sonraki 2 hafta içinde dişi fetusun tüm
hücrelerinde inaktivasyon tamamlanmıştır.
Xq13 de inaktivasyon merkezi başlangıç noktasıdır.
steroid sülfataz gibi bazı genler inaktivasyondan kaçarak aktif
X inaktivasyon merkezi
Xq13 bandında yerleşmiştir.
XIST (İnaktif X (Xi)- spesifik transkriptler) adı verilen
alışılmadık bir geni içerir. Bu gen X inaktivasyonu için
düzenleyici lokus görevi yapar.
Sadece inaktif X üzerindeki alelden ifadelenir.
Aktif X üzerindeki alel erkek ve dişi hücrelerde sessizdir.
Yokluğu durumunda X inaktivasyonu gerçekleşmez.
Ürünü Barr cisimciği ile kompleks oluşturan kodlayıcı
olmayan RNA’dır.
XX XX XX XX
Muhtamelen 4 hücreli evreden sonra X lerden biri seçilerek inaktive edilir
Yavru hücreler kaynaklandıkları hücrede seçilen X hangisi ise onu inaktive eder
Dozaj kompansasyonu, her hücrede ata hücresi ile aynı X aktif, fakat hiç birinde iki X birden aktif değil
Farklı hücre kümeleri, farklı aktif X kromozomu taşır
XXXX Hücre bölünmesi XX Zigot XX XXXX XX
1. Psödootozomal bölgede bulunan genler
2.Psödootozomal bölge
dışında kalıp Y üzerinde kopyası bulunmayan
genler
*steroid sülfataz geni
inaktivasyondan kaçarak aktif kalır
(mutasyon X geçişli iktiyozis)
3. Uzun ve kısa kolda bulunan psödootozomal bölge dışındaki homoloji gösteren genler.
Inaktivasyonun rastgele olmadığı durumlar:
1. X otozomal translokasyon varlığında, normal X inaktive olur.
2. Yapısal bozukluk içeren X kromozomu inaktive olur. 3. Ekstraembriyonik dokularda paternal X inaktive
olur.
geç replike olan inaktif X, Barr cisimciğini oluşturur. Her hücrede sahip olduğu X kromozomundan bir
TURNER SENDROMU
X kromozomunun monozomisi ile birliktedir
Doğumda ve puberteden önce belirgin fenotipleri nedeniyle saptanabilirler 45,X olgularının %94 intrauterin exitus %5 spontan abortus %1 canlı doğum 1/500 canlı kız doğum
Temel kusur mayotik kromozom ayrılamamasıdır %70 paternal kaynaklıdır
Kistik higroma
Ultrasonla saptanabilir! Streak gonad
Primer amenore en sık şikayet Zeka normaldir
Doğumda ayak üstünde belirgin Lenf ödem dikkati çeker
45 46 Mozaiklik
mayoz sırasında
veya zigot oluştuktan sonra olabilir
Turner sendromuyla birlikte bulunan karyotipler 45,X %50 45,X /46,XX %15 46,X,i(Xq) %15 45,X/46, X, i(Xq) %5 46,X,Xq- veya 46,X, Xp- %5 Diğer 45,X / ? %5 46,X,r(X) 46,i(Yq)
46,XY X del(X)(p1-pter) iX(q)
Xq-Ring X X(p21 23)
Klinik farklıklıklar kromozom kusuruna bağlıdır
46,X, del(Xp11): spontan mensturasyon(+) somatik bulgular hafiftir.
46,X,del(Xq): primer amenore, meme gelişimi geri over yetmezliği ile gider.
46,i(Xq): mozaik durumda saptanır Xp del gibidir.
46,X,i(Xp): 45,X bulgularıyla benzerlik gösterir
46,X,r(X): spontan abortuslarda sık rastlanır, genellikle mozaiktir.
Seyrek olarak 45,X erkek fenotipinde olabilir. 45,X/ 46,XY
SRY lokusunu taşıyan saptanamamış Y;otozom
translokasyonları sonucu gelişebilir.
Noonan Sendromu:
Fenotip erkek ,
Turner sendromu bulgularına sahip Karyotip 46,XY
47,XXX
1000 canlı kız doğumda bir görülür.
Çoğunlukla bir bulgu vermez ve bu nedenle tesadüfen
teşhis edilir.
% 5 vakada hafif mental retardasyon gözlenir. Bu etkilenim
sadece akademik hayatı etkileyecek düzeydedir. Nadiren 45-70 IQ arasında olgular gözlenir.
1/3 olguda POF gözlenebilir.
Gebe kaldıklarında %50 vakada yine sayısal seks
kromozom düzensizlikleri gözlenebilir.
Depresyon sıklığı artmıştır.
X sayısı arttıkça mental retardasyon ağırlaşır.
XXXXX vakaları fizik muayene ile Down sendromundan
FRAJİL X SENDROMU
Xq27 de yerleşik FMR1 genindeki CGG
tekrar artışına bağlı gelişir (dinamik
mutasyon)
Premutasyon (55-200 CGG) taşıyıcılığı
durumunda prematür ovaryen yetmezlik
bulunur.
Menapoz vakaların yarısından çoğunda 40
yaş öncesi görülür
KLINEFELTER SENDROMU
Fenotip erkek Karyotipte
En az bir Y, en az iki X vardır
Hipogonadizm bulgularının ortaya çıktığı pubertal döneme kadar normal olarak değerlendirilirler.
Uzun ve ince görünümlü, Unikoid yapılıdırlar(bacaklar daha uzun). Testisler küçük Azospermik Gonadotropinler yüksektir (FSH,LH) testosteron düşüktür (hipergonadotropik hipogonadizm) Jinekomasti %40 mikropenis
Toplumda 1/1000
Spontan abortuslarda 1/300 sıklıkta rastlanır Mozaikler dışında infertildirler
Fazla X kromozomu kaynağı
%47 maternal %53 paternal
Paternal 1. Mayoz hatası %50 Maternal 1. Mayoz hatası %30
2. Mayoz ve post zigotik ayrılma hatası(Jacops ve ark,1988)
%15 Mozaik yapı saptanır
Klinefelter Varyantları 1
48,XXXY: ağır mental retardasyon
49,XXXXY: Fraccaro sendromu fenotip Down S’ na benzer, ağır MR ve hipogonadizm bulunur.
1. ve 2. Mayozda birbirini izleyen ayrılamama sonucu oluşur
48,XXYY: hipogonadizm, ağır MR, agressivite Sıklık 1/25 000
Klinefelter Varyantları 2
46,XX: klinefelter sendromu bulguları
boy kısalığı
testiküler atrofi infertilite
46,XY(X kromatin negatif Klinefelter S) Del Castillo S. da denir
Hipogonadizm
47,XXY
48,XXXY
49,XXXXY
48,XXYY
46,XX
46,XY
47,XXY/46,XY
47,XYY sendromu
1.1/ 1000 Uzun boy
Antisosyal davranış bozukluğu
Suç işlemeye yatkınlık
Dış genital sistem gelişim bozukluğu
YY sperm oluşturan paternal 2. Mayoz ayrılma kusurudur.
48, XYYY Sendromu
Genital anomali Davranış bozukluğu Orta derecede MR
Hem testis hem de over bulunur.
Genellikle şüpeli dış genital görünüm saptanır. Tipik karyotip 46,XX
46,XY (%10)
XX/XY kimerizmi olabilir.
Gerçek Hermafroditizm
Yalancı Hermafroditizm
Yalnızca bir cinsiyete ait gonadal gelişim yanında
46,XY karyotipi taşır.
•Gonadotropinlere ait anormallikler
•Doğuştan testesteron biyosentez bozuklukları
(5 reduktaz enzim eksikliği)
•Androjene hedef dokuda duyarsızlık
Genetik olarak da klinik olarak da heterojendir.
Komplet androjen duyarsızlığı vardır. Androjen reseptör fonksiyonu bozuktur. Bu gen Xq13 de bulunur.
•X’ e bağlı resesif kalıtılır •Sıklık 1/20 000
•Karyotip 46,XY FENOTİP:
Normal kadın dış genital gelişimi Kör bir vajina bulunur
Uterus ve tubalar bulunmaz
Testisler genellikle karın içinde kalır (kriptoorşitizm) veya kasıkta yerleşir.
Çocukken geçirilmiş fıtık ameliyatı öyküsü tipiktir.
•Kortizol sentezinden sorumlu enzim defektleri Konjenital Adrenal Hiperplazi
•46,XX karyotipi taşır •Otozomal resesif
Overyan gelişim normaldir.
kız bebeklerin virilizasyonu gözlenir, bazen şüpeli genitalyaya neden olur.
En sık 21 OH laz enzim eksikliği bulunur.
Sıklık 1/12 500
Androjen sentezinin artışı klitoral büyüme, skrotum benzeri yapı oluşturan labial füzyon ile giden
virilizasyona neden olur.
Homozigot kız infantlarda şüpeli genitalya (ambigious genitale) bulunur.
Erkek infantlarda tanı gecikir. %25 basit virilizan tip
%75 tuz kaybettirici tip (tedavi edilmezse ölümcüldür)
Cinsiyet kromozom anormallikleri
Cinsiyet hastalık karyotip
Erkek Klinefelter 47,XXY
47,XYY sendromu 47,XYY
X ve Y kromozomu İçeren diğer
anormallikler
46,XX male 46,XX
Kadın Turner sendromu 45,X
46,X,i(xq) diğer delesyon
ve mozaik durumlar 47,XXX
XY female 46,XY