T.C.
EGE ÜNİVERSİTESİ TIP FAKÜLTESİ
ÇOCUK SAĞLIĞI VE HASTALIKLARI ANA BİLİM DALI PROF. DR. SAVAŞ KANSOY
ÇOCUKLUK ÇAĞI OTOİNFLAMATUAR
HASTALIKLARDA
GENOTİP-FENOTİP İLİŞKİSİ
UZMANLIK TEZİ
Dr. Gamze VURAN
DANIŞMAN Prof. Dr. Afig BERDELİ
ÖNSÖZ
Eğitimime değerli katkılarında dolayı, başta Anabilim Dalı Başkanımız Prof. Dr. Savaş KANSOY olmak üzere tüm Ege Üniversitesi Çocuk Sağlığı ve Hastalıkları AD Öğretim Üyeleri’ne,
Tez konusunun seçiminde yardımcı olan, tez çalışmam boyunca sahip olduğu bilgi, birikim ve görüşleriyle beni yönlendiren tez danışmanım Sayın Prof. Dr. Afig BERDELİ’ye,
Birlikte çalışmaktan zevk aldığım ve zorlu eğitim sürecini daha yaşanabilir kılan asistan arkadaşlarıma,
Tezimin istatistiklerini yapmamda yardımcı olan Biyoistatistik Anabilim Dalı’ndan Araştırma Görevlisi Hatice ULUER’e,
Tez hastalarımı çağırmamda, verilerimi toplamamda yardımcı olan başta Ceren YAŞAR olmak üzere tüm Moleküler Genetik Laboratuvar ekibine,
Sadece tezim süresince değil, yaşamımın her anında varlığını yanımda hissetmekten büyük mutluluk duyduğun eşim Özcan VURAN’a,
Her zaman yanımda olan ve desteklerini benden esirgemeyen AİLEME sonsuz teşekkürlerimi sunarım.
Dr. Gamze VURAN İzmir - 2017
ÖZET
ÇOCUKLUK ÇAĞI OTOİNFLAMATUAR HASTALIKLARDA GENOTİP-FENOTİP İLİŞKİSİ
Amaç: Ege Üniversitesi Tıp Fakültesi Çocuk Sağlığı ve Hastalıkları Ç. Romatoloji ve Genel Pediatri polikliniklerine Ailesel Akdeniz Ateşi (FMF) ve otoinflamatuar hastalık ön tanısıyla yönlendirilen, MEFV geninde mutasyon saptanmayan 40 olgunun diğer otoinflamatuar hastalıklar (Tümör Nekroz Faktör Reseptör 1 İlişkili Periyodik Sendrom (TRAPS), Hiperimmunglobulin D Sendromu (HIDS), Kriyopirin İlişkili Periyodik Sendrom (CAPS), Pyojenik artrit, Pyoderma Gangrenosum ve Akne Sendromu (PAPA) ) açısından yeni nesil dizileme (NGS) yöntemi ile değerlendirilmesi amaçlandı. Bu çalışma kesitsel ve kontrollü bir çalışmadır.
Gereç ve Yöntem: Çalışmaya dahil edilen 40 olgu ve ebeveynleri ile görüşülerek anket formu (hastalığın başlangıç yaşı, ataklara eşlik eden semptomlar, aile öyküsü, aldıkları tedavi, atak döneminde yapılan tetkik bilgilerini içeren) dolduruldu. Her olgudan 2 cc kan EDTA’lı tüpe alındı. Genetik analiz için Yeni Nesil Dizileme yöntemi ile kullanıldı. Çıkan sonuçlar veritabanlarına göre değerlendirildi. Araştırma verileri SSPS istatistik programında analiz edildi. P < 0.05 olduğu durumlar istatistik olarak anlamlı kabul edildi.
Bulgular: Çalışmaya dahil edilen 40 olgudan 16’sında mutasyon saptandı. Mutasyon pozitif olgularda hastalığın ortalama başlangıç yaşı 7.1 ± 4.6, ortalama tanıda gecikme yılı 4.1 ± 3.9, mutasyon negatif olgularda ise hastalığın ortalama başlangıç yaşı 5.2 ± 4.6 olarak saptandı. Mutasyon pozitif olgularda aile öyküsünün %37.5, mutasyon negatif olgularda %16.6 oranında olduğu görüldü. Her iki grupta da ataklara en sık eşlik eden semptomlar ateş, eklem ağrısı ve karın ağrısıydı. Mutasyon negatif olgularda aft ve makulopapüler döküntü mutasyon pozitif olgulara kıyasla anlamlı olarak daha yüksek saptandı (sırasıyla %41 - %0 p=0.003, %37 - %6 p=0.03) Mutasyon pozitif olgularda atak döneminde alınan akut faz reaktan değerlerinin mutasyon negatif olgulara göre istatiksel anlamlı olmasa da daha yüksek olduğu görüldü. NLRP3 gen mutasyonu saptanan 7 olgu CAPS olgu grubuna dahil edildi. 7 hastadan 5’inde Q703K, 1’inde V198M ve 1’inde I313V mutasyonu saptandı. Bu olgularda ortalama hastalık başlangıç yaşı 6.5 ± 3.8 idi. Ataklara en sık eşlik eden semptomlar ateş, artralji, karın ağrısı ve döküntüydü (sırasıyla %86, %71, %43 ve iii
%43). MVK geninde mutasyonu saptanan 6 olgu HIDS olgu grubuna dahil edildi. 6 hastadan 5’inde S52N, 1’inde V377I mutasyonu saptandı. Bu olgularda ortalama hastalık başlangıç yaşı 8.4 ± 5.1 idi. Ataklara en sık eşlik eden semptomlar artralji, karın ağrısı ve döküntüydü (sırasıyla %100, %50 ve %50). TNFRSF1A geninde mutasyonu saptanan 2 olgu TRAPS olgu grubuna dahil edildi. 2 hastadan 1’inde C52Y mutasyonu, 1’inde R92Q mutasyonu saptandı. Bu olgularda ortalama hastalık başlangıç yaşı 7.7 ± 8.8 idi. Ataklara en sık eşlik eden semptomlar ateş, karın ağrısı, artraljiydi (sırasıyla %100, %50, %50). PSTPIP1 geninde A372V mutasyonu saptanan 1 olgu PAPA olgu grubuna dahil edildi. Hastalık başlangıç yaşı 2 yaş olan bu olgumuzun ateş, karın ağrısı ve artraljinin eşlik ettiği 1 gün süren atakları mevcuttu. Tartışma ve Yorum: Bu çalışma Türkiye’de otoinflamatuar hastalıklarla ilgili yapılan kesitsel ve kontrollü ilk çalışmadır. Çalışmamızda saptanan mutasyondan 4’ü kesin patojenik (V377I, C52Y, I313V, A372V), 3’ü düşük penetranslı mutasyon (Q703K, V198M, R92Q), 1’i de patojenik önemi bilinmeyen (S52N) mutasyondur. Mutasyon pozitif hasta grubumuzda hastalık başlangıç yaşının literatürdeki olgulara göre daha geç olmasının, klinik bulgularının daha hafif olmasının, ciddi nörolojik tutulum ve kas iskelet sistemi tutulumu olmamasının, hastalarımızın büyük çoğunluğunda (12 hastada) düşük penetranslı mutasyonların saptanması ile ilişkili olduğu düşünülmüştür. Q703K mutasyonu saptanan 5 olgu ve S52N mutasyonu saptanan 5 olgu incelendiğinde, aynı mutasyona sahip olgularda hastalık başlangıç yaşlarının ve klinik semptomların birbirinden farklı olduğu izlenmiş, mutasyonların değişken penetransı ve oluşturdukları heterojen fenotip nedeniyle aynı mutasyonun farklı olgularda farklı klinik tablo (asemptomatik - ağır atak) yapabileceği görülmüştür. Tekrarlayan ateş, artralji, karın ağrısı yakınmaları ile başvuran FMF öntanısıyla refere edilen ancak MEFV geninde mutasyon saptanmayan olgularda düşük insidans oranına rağmen diğer otoinflamatuar hastalıklar göz önünde bulundurulmalı ve klinik şüphe halinde buna yönelik ileri genetik inceleme yapılmalıdır.
SUMMARY
GENOTYPE-PHENOTYPE RELATIONSHIP IN CHILDHOOD AUTOINFLAMMATORY DISEASES
Objective: 40 cases with no mutation in MEFV gene, directed with FMF and autoinflammatory disease pre-diagnosis to Pediatric Rheumotology and General Pediatrics polyclinics of Pediatrics in Medical School of Aegean University have been aimed to be assessed with New Generation Sequencing (NGS) in terms of other auto-inflammatory diseases (Tumor Necrosis Factor 1 Associated Periodic System (TRAPS), Hyperimmunoglobulobin D Syndrome (HIDS), Cryopyrine Associated Periodic Syndrome (CAPS), Pyogenic Arthritis, Pyoderma Gangrenosum and Acne Syndrome (PAPA) ). This study is a cross-sectional and controlled study.
Material and Method: 40 cases and their parents involved in the study were interviewed and survey form was filled in (including onset age of disease, symptoms accompanying the attacks, family history, received treatment, information of examinations performed in attack period). 2 cc blood was taken from each case into EDTA tube. New Generation Sequencing method was used for genetic analysis. The obtained results were assessed according to databases. The research data were analyzed in SSPS statistical program. The conditions of P < 0.05 were accepted as statistically significant.
Findings: Mutation was detected in 16 of the 40 cases involved in the study. In cases with mutation positive, the average age of the disease onset was detected to be 7.1 ± 4.6, average year of delayed diagnosis to be 4.1 ± 3.9, in cases with mutation negative, the average age of disease onset was detected to be 5.2 ± 4.6. Family history was observed to be 37.5% in mutation positive cases and to be 16.6% in mutation negative cases. The most common symptoms accompanying the attacks in each group were fever, arthralgia and abdominal pain. Aphtha and maculopapular rash were determined to be significantly higher in mutation negative cases than mutation positive cases (p = 0.003, p = 0.03).
Acute phase reactant levels taken in the attack period in mutation positive cases were seen to be higher than mutation negative cases even though not statistically significant. 7 cases detected with NLRP3 gene mutation were included in CAPS case group. In 5 of 7 patients Q703K mutation, in 1 of them V198M and in 1 of them I313V mutation v
were detected. The average age of disease onset was 6.5 ± 3.8 in these cases. The most common symptoms accompanying the attacks were fever, arthralgia, abdominal pain, and rash (86%, 71%, 43%, and 43%, respectively). 6 cases detected with MVK gene mutation were included in HIDS case group. S52N mutation was found in 5 and V377I mutation was found in 1 of 6 patients. The average age of disease onset in these cases was 8.4 ± 5.1. The most common symptoms accompanying the attacks were arthralgia, gastralgia and rash (100%, 50% and 50%, respectively). 2 cases detected with TNFRSF1A gene mutation were included in TRAPS case group. In one of the two patients C52Y mutation and in the other one R92Q mutation were found out. The average age of disease onset in these cases were 7.7 ± 8.8. The most common symptoms accompanying the attacks were fever, abdominal pain and arthralgia (100%, 50%, 50% respectively). 1 cases detected with A372V mutation in PSTPIP1 gene was
included in PAPA case group. This case of ours the disease onset of whom was 2 years had one
day lasting attacks accompanied by fever, abdominal pain, arthralgia.
Discussion and conclusion: This study is the first cross sectional and controlled study made on auto-inflammatory diseases in Turkey. 4 of the mutations found in our study were definite pathogenic (V377I, C52Y, I313V, A372V), 3 of them were low penetrance mutations (Q703K, V198M, R92Q), and 1 of them was the mutation with uncertain significance (S52N). The fact that in our mutation positive patient group, the disease onset age was older than the cases in literature, their clinical findings were lighter, and there were not serious neurological involvement and musculoskeletal involvement was thought to be associated with the fact that low penetrance mutations were found in most of our patients (12 patients). In examining 5 cases detected with Q703K mutation and 5 cases detected with S52N mutation, the disease onset age and clinical symptoms in cases with the same mutation were observed to be different from one another, and mutations were observed to have changeable penetrance, and the same mutation were observed to cause different clinical picture (asymptomatic - severe attack) in different cases because of heterogeneous phenotype they created. In cases having applied with repetitive fever, arthralgia and abdominal pain complaints, referred with FMF pre-diagnosis, but not detected with mutation in MEFV gene, other auto-inflammatory diseases must be considered despite the low incidence rate, and prospective genetic examination must be carried out for it in the event of clinical suspect.
İÇİNDEKİLER ÖNSÖZ ... ii ÖZET... iii SUMMARY ... v TABLO LİSTESİ ... x ŞEKİL LİSTESİ... xi KISALTMALAR ...xii 1. GİRİŞ VE AMAÇ... 1 2. GENEL BİLGİLER ... 2 2.1. Tanım ... 2 2.2. Tarihçe ... 2 2.3. Otoinflamatuar Hastalık ... 3 2.3.1. Epidemiyoloji ... 3 2.3.2. Etyopatogenez ... 3 2.3.2.1. İnflamazom ... 5 2.3.2.2. Noncanonical İnflamazom ... 10
2.3.2.3. ASC ve İnflamasyonun/inflamazomun yayılımı ... 11
2.4. Ailevi Akdeniz Ateşi (FMF) ... 11
2.4.1. Epidemiyoloji ... 12
2.4.2. Ailevi Akdeniz Ateşi’nde Genetik ... 12
2.4.3. Patogenez ... 14
2.4.4. Klinik ... 15
2.4.5. Uzun Dönem Komplikasyonları ... 17
2.4.6. Laboratuvar ... 19
2.4.7. Tanı ... 19
2.4.7.1. Genetik Tanı ... 19
2.4.7.2. Ayırıcı tanı ... 20
2.4.8. Tedavi ... 20
2.5. Tümör Nekroz Faktör Reseptör 1 İlişkili Periyodik Sendrom (TRAPS) ... 22
2.5.1. Epidemiyoloji ... 22
2.5.2. Tümör Nekroz Faktör İlişkili Periyoidk Sendrom’da Genetik ... 23
2.5.3. Patogenez ... 24 2.5.4. Klinik ... 26 2.5.5. Laboratuvar ... 27 2.5.6. Tanı ... 28 2.5.6.1. Ayırıcı Tanı ... 28 2.5.7. Tedavi ... 28
2.6. Hiperimmunglobulin D Sendromu (HIDS) ... 28
2.6.1. Epidemiyoloji ... 29
2.6.2. Hiperimmunglobulin D Sendromu’nda Genetik ... 29
2.6.3. Patogenez ... 30
2.6.3.1. Mevalonat yolağı ... 30
2.6.3.2. Genetik defekt ... 32
2.6.3.3. İnflamazom aktivasyonu ve IL-1B sekresyonu ... 32
2.6.4. Klinik ... 33 2.6.5. Laboratuvar ... 34 2.6.9. Tanı ... 34 2.6.9.1. Genetik Tanı ... 34 2.6.9.2. Ayırıcı tanı ... 35 2.6.10. Tedavi ... 35 2.6.10.1. Semptomatik tedavi ... 35 2.6.10.2. Önleyici tedavi ... 36
2.7. Kriyopirin İlişkili Periyodik Sendromlar (CAPS) ... 36
2.7.1. Epidemiyoloji ... 37
2.7.2. Kriyopirin ilişkili Periyodik Sendrom’da Genetik ... 37
2.7.3. Patofizyoloji ... 38
2.7.4. Klinik ... 38
2.7.4.1. Ailesel Soğuk Otoinflamatuar Hastalık (FCAS) ... 38
2.7.4.2. Muckle-wells sendromu (MWS) ... 39
2.7.4.3. Erken başlangıçlı multisistem otoinflamatuar hastalık (CINCA/NOMID) ... 39 2.7.5. Laboratuvar ... 39 2.7.6. Tanı ... 40 2.7.6.1. Ayırıcı tanı ... 40 2.7.7. Tedavi ... 40 viii
2.8. Periyodik Ateş, Aftöz Stomatit, Farenjit, Adenit (PFAPA) Sendromu ... 41 2.8.1. Epidemiyoloji ... 41 2.8.2. Genetik ... 41 2.8.3. Patogenez ... 41 2.8.4. Klinik ... 42 2.8.5. Laboratuvar ... 42 2.8.6. Tanı ... 43 2.8.6.1. Ayırıcı tanı ... 43 2.8.7. Tedavi ... 43
2.9. Piyojenik Artrit, Piyoderma Gangrenozum ve Akne Sendromu (PAPA) ... 44
3. GEREÇ VE YÖNTEM ... 47
3.1. Çalışmanın Tasarımı ... 47
3.1.1. Yeni Nesil Dizileme (Next Generation Sekanslama) ... 47
3.1.1.1. DNA İzolasyon ... 47
3.1.1.2. DNA'nın Kontrolü ... 48
3.1.1.3. Yeni Nesil Dizileme Yöntemi ... 48
3.2. Veri Analizinde Kullanılan İstatiksel Yöntemler ... 49
4. BULGULAR ... 50
5. TARTIŞMA ... 58
6. SONUÇ VE ÖNERİLER ... 68
KAYNAKLAR ... 70
EKLER ... 94
Ek 1: Etik Kurul Onayı ... 94
Ek 2: Olgu Rapor Formu ... 97
EK 3: Özgeçmiş Formu ... 102
TABLO LİSTESİ
Tablo 2.1. Hastalıklara karşı inflamazomların koruyucu ve zararlı etkileri ... 11 Tablo 2.2. FMF tanı kriterleri ... 19 Tablo 4.1. Çalışmaya dahil edilen hastalara ait demografik veriler ... 50 Tablo 4.2. Çalışmaya dahil edilen hastaların atak sırasında izlenen klinik
bulgularına ait veriler ... 53 Tablo 4.3. Çalışmaya dahil edilen hastaların atak dönemine ait laboratuvar
verileri ... 54 Tablo 4.4. Çalışmaya dahil edilen hastaların almış oldukları medikal tedaviler .... 55 Tablo 4.5. Kriyopirin ilişkili periyodik sendromlu (CAPS) olgularda
genotip-fenotip ilişkisi ... 55 Tablo 4.6. Hiperimmunglobulin D sendromlu (HIDS) olgularda genotip-fenotip
ilişkisi... 56 Tablo 4.7. Tümör nekroz faktör reseptör 1 ilişkili periyodik sendromlu (TRAPS)
olgularda genotip-fenotip ilişkisi ... 57 Tablo 4.8. Pyojenik artrit, pyoderma gangrenosum ve akne (PAPA) sendromlu
olgularda genotip- fenotip ilişkisi ... 57
ŞEKİL LİSTESİ
Şekil 2.1. Genel inflamazom yapısı ve fonksiyonları ...6
Şekil 2.2. İnflamazomlar ...7
Şekil 2.3. NLRP3 inflamazom aktivasyonu için iki aşamalı sinyal ...8
Şekil 2.4. Pirin aksiyon mekanizmaları... 10
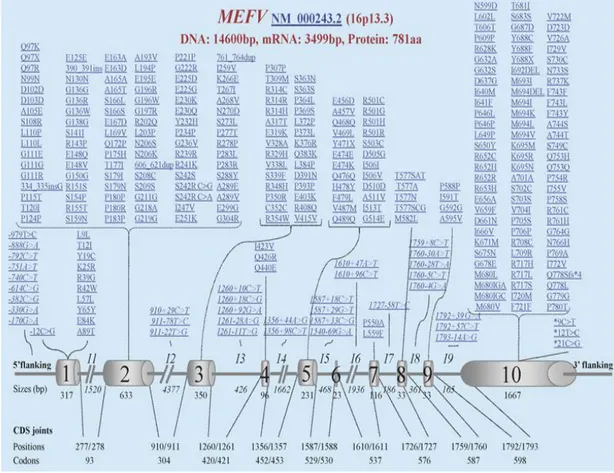
Şekil 2.5. İnfevers veri tabanında MEFV geninde saptanan mutasyonlar ... 12
Şekil 2.6. MEFV geni ve kodladığı pirin proteini ... 14
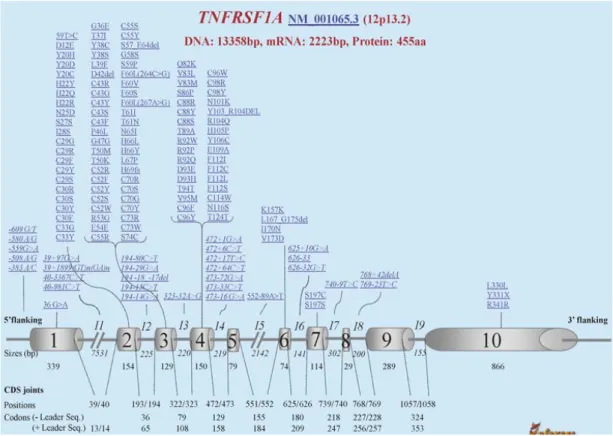
Şekil 2.7. İnfevers veritabanına göre TNFRSF1A geninde saptanan mutasyonlar ... 22
Şekil 2.8. TNFRSF1A geni ve kodladığı TNF alfa reseptör proteini ... 23
Şekil 2.9. TRAPS patogenezi ... 26
Şekil 2.10. İnfevers veritabanına göre MVK geninde saptanan mutasyonlar ... 30
Şekil 2.11. MVK geni ve kodladığı protein ... 30
Şekil 2.12. Mevalonat yolağının ürünleri. Mevalonat yolunun şematik gösterimi . 31 Şekil 2.13. İnfevers veritabanına göre NLRP3 geninde saptanan mutasyonlar ... 37
Şekil 2.14. NLRP3 geni ve kodladığı kryopirin proteini ... 38
Şekil 2.15. İnfevers veritabanına göre PSTPIP1 geninde saptanan mutasyonlar .... 45
Şekil 2.16. PSTPIP1 geni ve kodladığı protein ... 45
KISALTMALAR
ALR Absent in melanoma benzeri reseptör (Absent in melanoma Like Receptor)
ASC Kaspaz tanıma alanı içeren apoptoz ile ilişkili benek benzeri protein (Apoptosis-associated Speck-like Protein containing a CARD)
CANDLE Lipodistrofinin eşlik ettiği kronik atipik nötrofilik dermatoz ve ateş sendromu (Chronic Atypical Neutrophilic Dermatosis with Lipodystrophy and Elevated Temperature Syndrome)
CAPS Kriyopirin ilişkili Periyodik Ateş sendromları (Cryopyrin Associated Periodic Fever Syndrome)
CINCA Kronik İnfantil Nörolojik Kutanöz Artiküler sendrom (Chronic Infantile Neurological Cutaneous and Articular)
CRMO Kronik Rekürren Multifokal Osteomyelit Sendromu (Chronic Recurren Multifocal Osteomyelitis)
DAMP Hasar ilişkili moleküler patern (Damage Associated Molecular Pattern) FCAS Ailesel soğuk otoinflamatuar hastalık (Familial Cold
Autoinflammatory Syndrome)
FLIP FLICE-benzeri inhibitör protein (FLICE-like İnhibitory Protein) FMF Ailevi Akdeniz Ateşi (Familial Mediterranean Fever)
FPP Farnesil pirofosfat GGPP Geranilgeranil pirofosfat
HIDS Hiperimmunglobulin D Sendromu HMG-CoA 3- hidroksi-3-metilglutaril koenzim A IL-1 İnterlökin 1
IPP İzopentenil pirofosfat
IRF İnterferon düzenleyici gen (Interferon Regulatory Factor) LPS Lipopolisakkarit
LRR Lösinden zengin tekrarlar (Leucine Rich Repeat) MKD Mevalonat kinaz enzim defekti
MWS Muckle-Wells sendromu NF-Kb Nükleer faktör kappa B
NOD Nükleotid bağlayıcı oligomerizasyon domain (Nucleotide Binding Oligomerization Domain)
NLR NOD benzeri reseptör (NOD Like Receptor) NK Doğal öldürücü hücre (Natural Killer)
PAMP Patojen ilişkili moleküler patern (Pathogen Associated Molecular Pattern )
PAPA Pyojenik Artrit, Pyoderma Gangrenozum ve Akne Sendromu PCR Polimeraz zincir reaksiyonu
PFAPA Periyodik Ateş, Aftöz Stomatit, Farenjit, Adenit sendromu (Periodic Fever, Apthous Stomatitis, Pharyngitis, Adenitis Syndrome)
PLAID Pre-ligand montaj alanı (Pre-ligand Binding Assembly Domain) PBMC Periferik kan mononükleer hücreleri
PRR Patern tanıma reseptörleri (Patern Recognition Receptor) RIG Retinoik asit indüklenebilir gen (Retinoic Acid Inducible Gene) SAPHO Sinovit, Akne, Püstülozis, Hiperostozis ve Osteitis sendromu TIP Tioredoksin etkileşim proteini (Tioredoxin Interacting Protein) TLR Toll benzeri reseptör (Toll Like Receptor)
TNF Tümör nekroz faktör
TRADD TNF reseptör ilişkili ölüm bölgesi proteini (TNF receptor associated death domain)
TRAF TNF reseptör ilişkili faktör (TNF receptor associated factor) UPR Katlanmamış protein yanıtı (Unfolded Protein Response) RIP Reseptör etkileşim proteini (Receptor Interacting Protein)
TRAPS Tümör Nekroz Faktör ilişkili Periyodik Ateş Sendromu (Tumor Necrosis Factor Receptor 1 Associated Periodic Syndrome)
1. GİRİŞ VE AMAÇ
Otoinflamatuar hastalıklar; doğal immun sistem regülasyonundaki yetersizlik sonucu oluşan, tekrarlayan sistemik inflamatuar ataklarla karakterize nadir görülen bir grup hastalıktır. Tekrarlayan ateş, döküntü, lenfadenopati, serozit (peritonit, plörit) ve kas-iskelet sistemi gibi farklı sistemlerin tutulumu gözlenmektedir. Ataklar sırasında veya ataklar arasındaki dönemde otoantikor veya antijene spesifik T hücresinin gözlenmemesi, bu hastalıkları otoimmun hastalık grubundan ayırmaktadır. Bu hastalıklar çoğunlukla hayatın ilk saatleri - ilk dekatı içerisinde başlamaktadır. Nadir görüldüğü, çok bilinmediği ve farklı klinik bulgularla prezente oldukları için geç tanı almaktadırlar. Tanıda gecikme tekrarlayan ataklar sonucu kalıcı eklem sekellerinin oluşmasına neden olmaktadır. En sık görüleni ve bilineni Ailevi Akdeniz Ateşi (FMF) olmakla birlikte Tümör Nekrozis Faktör Reseptör 1 İlişkili Periyodik Sendrom (TRAPS), Hiperimmunglobulin D Sendromu (HIDS), Kriyopirin İlişkili Periyodik sendromlar (CAPS), Periodik Ateş, Aftöz Stomatit, Farenjitis, Adenitis (PFAPA) Sendromu bu grup hastalıklardandır.
Son yıllarda genetik analiz yöntemlerinin gelişmesi (Yeni Nesil Dizileme yönteminin kullanıma girmesiyle) ve otoinflamatuar hastalıklarla ilgili bilgi ve farkındalığın artmasıyla birlikte bu hastalıklarda tanı oranı artmıştır. Artan hasta sayısı ile birlikte mutasyonlar ve oluşturdukları klinik tabloyu inceleyen genotip-fenotip çalışmaları da artış göstermiştir.
Bu çalışma 2013-2017 tarihleri arasında tekrarlayan inflamatuar atakları olan, FMF ya da otoinflamatuar hastalık öntanılarıyla Ege Üniversitesi Çocuk Romatoloji ve Genel Pediatri polikliniklerine yönlendirilen, yapılan genetik analiz sonucu MEFV geninde mutasyon saptanmayan olgularda diğer otoinflamatuar hastalıkların (TRAPS, HIDS, CAPS Pyojenik artrit, pyoderma gangrenozum ve artrit (PAPA) sendromu) incelenmesini amaçlayan kesitsel ve kontrollü bir çalışmadır. Öncelikle mutasyon saptanan olguların klinikleri ile beraber değerlendirilerek genotip-fenotip ilişkisinin incelenmesi amaçlanmıştır. Otoinflamatuar hastalıklarla ilgili farkındalığın arttırılması bu sayede erken tanı ve hastalığa spesifik tedavinin erken dönemde başlatılmasının sağlanması da bu çalışmanın ikincil amacını oluşturmaktadır.
2.GENEL BİLGİLER 2.1. Tanım
Otoinflamatuar hastalıklar, herhangi bir patojen uyarısı olmaksızın tekrarlayan inflamatuar ataklarla seyreden bir grup hastalıktır. Doğal immun sistem regülasyonundaki yetersizlik sonucu oluşan bu hastalıklara eşlik eden yüksek titrede otoantikor ve otoreaktif T hücrelerin olmaması ile otoimmun hastalıklardan ayrılmaktadır. Bu hastalıklar tekrarlayan ateş ve eşlik eden deri, seröz zar, eklem, gastrointestinal sistem ve santral sinir sistemi bulguları ile seyretmektedir.
Periyodik ateş sendromları otoinflamatuar hastalıkların bir alt grubudur. Ailevi Akdeniz Ateşi (FMF), Tümör Nekroz Faktör Reseptör 1 ilişkili Periyodik Ateş Sendrom (TRAPS), Hiperimmunglobulin D Sendromu (HIDS), Kriyopirin ilişkili Periyodik Ateş Sendromları (CAPS) bu grup hastalıklardandır. Bu grup aynı zamanda monogenik otoinflamatuar hastalık grubuna girmektedir. Diğer monogenik otoinflamatuar hastalıklar; Majeed sendromu, Pyojenik Artrit, Pyoderma Gangrenozum ve Akne Sendromu (PAPA), Blau Sendromu, NALP12- İlişkili Periyodik Ateş Sendromu’dur. Poligenik otoinflamatuar hastalıklar; Periyodik Ateş, Aftöz Stomatit, Farenjit, Adenit sendromu (PFAPA), Behçet Hastalığı, Crohn Hastalığı, Still hastalığı, Shnitzler Sendromu, Sweet Sendromu, Kronik Rekürren Multifokal Osteomyelit (CRMO) Sendromu, Sinovit, Akne, Püstülozis, Hiperostozis ve Osteitis (SAPHO) sendromu’dur.
2.2. Tarihçe
“Periyodik hastalık” terimi ilk kez 1948 yılında Hobart Reimann tarafından benign paroksismal peritonit, periyodik ateş, siklik nötropeni, intermittant artraljinin görüldüğü 6 hastada bir sendrom olarak kullanıldı [1]. 1958 yılında Heller ve arkadaşları otozomal resesif kalıtılan ve Akdeniz kökenli olgularda daha sık görülen bu sendromu Ailevi Akdeniz Ateşi (FMF) olarak tanımladı [2]. İlerleyen yıllarda otozomal resesif ve otozomal dominant kalıtım gösteren, tekrarlayan ateşle seyreden diğer genetik sendromların da tanımlanmasıyla bu grup hastalıklar “Herediter Periyodik Ateş Sendromları” başlığı altında toplandı.
Otozomal dominant kalıtım gösteren ve ilk kez İrlandalı bir ailede saptanması nedeniyle de “Ailesel Hibernian Ateş” olarak adlandırılan TRAPS kliniği ilk kez 1982’de bir olguda bildirilmiştir [3]. Ailesel soğuk otoinflamatuar hastalık (FCAS) kliniği ilk kez 1940 yılında bir olguda [4], ürtiker, sağırlık ve amiloidozis ile karakterize Muckle-Wells sendromu (MWS) klinik tablosu 1962 yılında [5], Kronik İnfantil Nörolojik Kutanöz Artiküler sendrom (CINCA) klinik tablosu ilk kez 1981 yılında bir olguda bildirilmiştir [6]. Başlangıçta ayrı ayrı hastalıklar olarak değerlendirilen FCAS, MWS, CINCA sendromları 2001-2002 yıllarında CIAS1/NLRP3 gen mutasyonlarının bu üç hastalığın da sebebi olduğu gösterilmesi üzerine Kriyopirin İlişkili Periyodik Sendromlar (CAPS) olarak aynı başlık altında toplanmıştır [7-8]. Otozomal resesif kalıtılan ve tekrarlayan ateş, lenfadenopati, karın ağrısı ve raş ile karakterize Hiperimmunglobulin D Sendromu (HIDS) klinik tablosu ilk kez 1984 yılında bir olguda bildirilmiştir [9]. Herediter Periyodik Ateş sendromlarına neden olan genetik mutasyonların saptanmaya başlanması, bu hastalıklarda yüksek titrede otoantikor ve antijen spesifik T hücre görülmemesi üzerine otoimmun hastalıklardan ayırmak için “Otoinflamatuar Hastalık” terimi 1999 yılında kullanılmaya başlanmıştır [10-11].
2.3. Otoinflamatuar Hastalık 2.3.1. Epidemiyoloji
Otoinflamatuar hastalıklar, FMF ve PFAPA dışında nadir görülen hastalıklardır. FMF prevelansının Türk, Ermeni, Arap toplumlarında 1/200-1/1000 olduğu belirtilmektedir. HIDS ve TRAPS prevalansı net bilinmemekle birlikte Fransa’da yapılan bir çalışmada CAPS hesaplanmış prevalansı 1/360.000 olarak bildirilmiştir [12].
2.3.2. Etyopatogenez
İnflamasyon konak için zararlı uyaranlara karşı (patojen, irritan maddeler vb.) doğal immun sistemin verdiği koruyucu bir yanıttır. İnflamasyon yanıtı yetersiz olduğunda patojenlerin persistan enfeksiyonu, aşırı olduğunda ise kronik yada sistemik inflamatuar hastalıklar meydana gelmektedir [13]. Bağışıklık sistemi doğal
immun sistem ve adaptif immun sistem olmak üzere iki bölümden oluşmaktadır. Doğal immun sistem hücre içi hasarlanmada ve dış patojenlere karşı savunmada ilk non-spesifik hücre mekanizmasıdır. Doğal immun sistem bileşenlerini mekanik bariyer olan epitel, endotel, doğal öldürücü (NK) hücreler, mast hücresi, makrofaj, eozinofil, bazofil, nötrofil, dentritik hücreler, kompleman sistemi, inflamatuar sinyallerin iletimini sağlayan IL-1, TNF alfa gibi sitokinler oluşturmaktadır.
Enfeksiyöz patojenleri ve hücre içi tehlike sinyallerini algılayarak hızlıca proinflamatuar sitokinlerin salınımını sağlayan doğal immun sistem, germline’da kodlanan ve patojen ilişkili moleküler paternleri (PAMP) ve endojen, nonenfeksiyöz tehlike sinyallerini, hasar ilişkili moleküler paternleri (DAMP) tanıyan reseptörlere sahiptir. Bu reseptörler patern tanıma reseptörleri (PRR) olarak adlandırılmaktadır. Bu reseptörler; hücre membranı ve endozomlarda yer alan Toll benzeri reseptörler (TLR), sitoplazmik nükleotid bağlayıcı oligomerizasyon bölgesi (NOD) benzeri reseptörler (NLR), bir pirin bölgesi ve intraselüler mikrobiyal DNA’nın tanındığı DNA bağlayıcı HIN bölgesine sahip absent in melanoma 2 (AIM2) benzeri reseptörler (ALR), intraselüler yerleşimli ve antiviral immunitede önemli RIG-1 (retinoik asit indüklenebilir gen-1) benzeri reseptörler olmak üzere 5 farklı tiptedir [14].
Toll benzeri reseptörler yanlızca mikroorganizmaya ait yapıları tanırlar. TLR4 lipopolisakkaride; TLR2 peptidoglikan, lipoteikoik asite; TLR5 flagelline; TLR3 çift zincirli RNA’ya TLR7 ve 8 tek zincirli RNA’ya; TLR9 metillenmiş DNA’ya duyarlıdır [15]. Bu reseptörler farklı hücre içi sinyal yolaklarını aktive ederek inflamasyonu tetiklerler. Örneğin TLR4’ün uyarılması ile hücre içi MAP kinazlar, NFkB (nükleer faktör kappa), IRF (interferon düzenleyici faktör) aktive olur ve ilk ikisi ile inflamatuar sitokin üretimini sağlayan genlerin transkripsiyonu, 3.yolak ile tip 1 interferon üretimini sağlayan genlerin transkripsiyonunun başlatılması sağlanır.
NOD benzeri reseptörler; C-terminal LRR (lösinden zengin tekrarlar) bölgesi, santral nükleotid bağlayıcı bölge (NACHT), N terminal efektör bölge olmak üzere 3 bölümden oluşmaktadır [15-16]. C-terminal bölgedeki lösinden zengin tekrarlar (LRR) PAMP’lar için bir sensör görevi görmektedir. N-terminal bölge diğer proteinlerle etkileşerek efektör fonksiyonları gerçekleştirir. NACHT alanı ATPaz aktivitesine sahiptir ve oligomerizayonu sağlamaktadır. NOD benzeri reseptör ailesini sınıflandırmada kullanılan 4 N-terminal bölge mevcuttur; asidik transaktivasyon bölgesi (NLRA), baculoviral inhibitör tekrar bölgesi (NLRB), kaspaz aktive edici bölgesi (CARD; NLRC), pirin bölgesi (NLRP).
NLR'ler, enfeksiyöz patojenleri (peptidoglikan, flagellin, viral RNA, fungal hifler vb.), konakçı hücrelerindeki enfeksiyöz olmayan ligandları (ATP, kolestrol kristalleri, ürik asit vb.) ve çevresel kaynaklardaki ligandları (asbest, silika, alaşım partikülleri, UV radyasyonu vb.) tanıyarak inflamatuar yanıtı aktive etmektedir. Aktive edilmiş NLR'ler, dört ayrı fonksiyon göstermektedir; inflamazom oluşumu, sinyal iletimi, transkripsiyon aktivasyonu ve otofaji [17].
PAMP ve DAMP’lar tarafından uyarılan NOD-benzeri reseptörler pro-IL-1β ve pro-IL-18 salınımını sağlamaktadır. Salgılan bu inaktif sitokinlerin aktif hale gelmesi için kaspazlara ihtiyaç vardır. Uyarının çeşidine göre NOD-benzeri reseptör ailesinin bir üyesi oligomerleşir ve bir multiprotein kompleks olan inflamazomu oluşturarak kaspaz-1’i aktif hale getirir.
2.3.2.1. İnflamazom
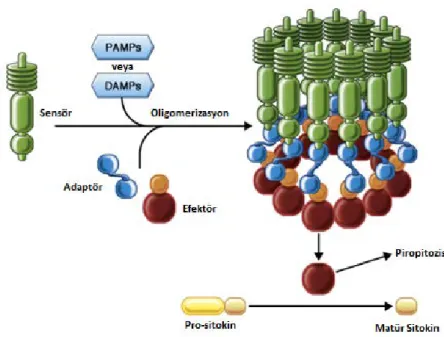
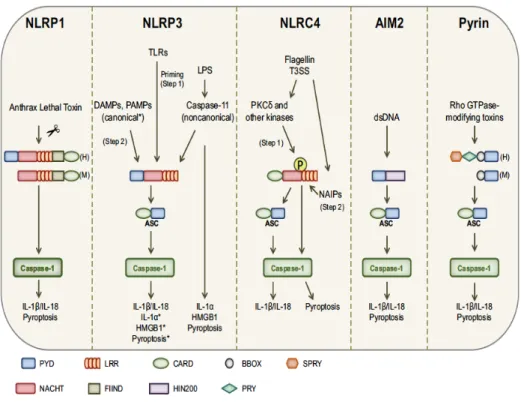
İnflamazomlar bir sensör protein (NOD-benzeri reseptör, AIM2 reseptör v.b.), bir adaptör protein (ASC- kaspaz tanıma alanı içeren apoptoz ile ilişkili benek benzeri protein) ve bir proinflamatuar kaspaz (kaspaz-1)’dan oluşmaktadır [18]. PAMP ve DAMP’lar tarafından inflamazomların uyarılması ile kaspaz-1 aktivasyonu gerçekleşmektedir [19]. Kaspaz 1’in aktive olmasıyla proinflamatuar sitokinler olan pro-IL-1β ve pro-IL-18 proteolitik ayrılma ile aktif IL-1β ve IL-18 haline getirilmektedir. Ayrıca inflamazomun aktivasyonu ile piroptozis de (patojenle enfekte hücrelerin ölümü) gerçekleşmektedir. İnflamazom yapısı Şekil 2.1’de, inflamazomları tetikleyen faktörler Şekil 2.2’de gösterilmektedir.
Şekil 2.1. Genel inflamazom yapısı ve fonksiyonları. Multimerik protein kompleksleri olan
inflamazomlar bir sensör protein, bir efektör ve sıklıkla bir adaptör poretin içerirler. DAMP ve PAMP’lar oligomerizasyonu tetikleyerek efektörü aktive eder ve hem prositokinlerin maturasyonu hem de piropitozisi sağlar.
İnflamazomlar içerdikleri sensör proteine göre isimlendirilmektedir. Çoğunlukla NOD-benzeri reseptör ailesinden bir sensör protein içerirler ve NLRP1, NLRP3, NLRC4 bunlara örnektir [20-21]. AIM2 ve pirin inflamazomu da NOD-benzeri reseptör içermeyen inflamazomlardır.
- NLRP1 inflamazomu: Martinon ve ark. tarafından tanımlanan NLRP1 inflamazomu ilk tanımlanan inflamazomdur [22]. İnsanlarda gram negatif ve gram pozitif bakterilerin peptidoglikan tabakası, muramil dipeptit (MDP), farelerde Basillus anthracis’in letal toksini NLRP1 inflamazomunu aktive etmektedir [23]. İnsanlarda pirin ve CARD domaini içeren bir NLRP1 geni vardır. MDP’lerin uyarısı ile NLRP1 inflamazomu oligomerize olarak kaspaz-1 aktivasyonunu sağlar ve inflamatuar sitokinler üretilir.
Şekil 2.2. İnflamazomlar: Tetikleyici faktörlerin etkisiyle oluşan inflamazomlar kaspaz-1’i aktive
ederek pro IL-1β ve pro-IL-18 üretimini arttırır ve piropitozisi tetikler. NLRP1 inflamazomu Basillus anthracis’in letal toksini tarafından tarafından aktive edilir. Canonical NLRP3 inflamazomu DAMP ve PAMP’lar tarafından uyarılmaktadır. Noncanonical NLRP3 inflamazomunda LPS‘ler kaspaz 11‘i aktive etmektedir. Bakteriyel flajellin ilk olarak protein kinazları aktive etmekte ardından NLRC4 fosforile olarak inflamazom formasyonunu oluşturmaktadır. AIM2 inflamazomu sitozolik dsDNA tarafından, pirin inflamazomu bakteriler tarafından yapılan Rho GTPaz üzerindeki modifikasyonlar ile uyarılmaktadır [24].
- NLRC4 inflamazomu: NLRC4 inflamazomu Legionella pneumophila, Pseudomonas aeruginosa, Salmonella typhimurium ve Shigella flexneri tarafından uyarılmaktadır [25-30]. Duyarlı olduğu bakteriyel flajellin ve bakteriyel tip 3 sekresyon sistemi direk olarak NAIP’e (NLR ailesi apoptozis inhibitör protein) bağlanır ve NLRC4 inflamazomu aktive ederek inflamasyonu başlatır.
- NLRP3 inflamazomu: NLRP3, NOD-benzeri reseptör ailesinin bugüne kadar üzerine en çok çalışılan üyesidir. NLRP3 myeloid hücrelerde düşük seviyede bulunmaktadır. Lipopolisakkaritler Toll-benzeri reseptörleri uyararak NF-kB bağımlı inflamatuar sitokinlerin salınımını sağlamaktadır. Böylece NLRP3 transkripsiyonun artması sağlanmaktadır [31]. “1. Sinyal“ olarak adlandırılan bu basamak pro-IL-1β transkripsiyonun artması ve NLRP3 inflamazomunun 2. sinyale cevap verir hale gelmesi için gereklidir. NLRP3 inflamazomu için tetikleyici faktörler olarak
PAMP’lar, bakteriyel, viral, protozoal, fungal toksinler ve DAMP’lar (ATP, ürik asit kristalleri, amiloid-β fibriller ) yer almaktadır [32-36].
Şekil 2.3. NLRP3 inflamazom aktivasyonu için iki aşamalı sinyal [37]. NLRP3, ASC ve pro-kaspaz 1’den oluşan NLRP3 inflamazomunun aktivasyonu, iki aşamalı sinyallerle sıkı bir şekilde düzenlenir. LPS gibi ilk başlatma sinyali, transkripsiyon faktörü NF-κB'nin aktivasyonu ile inflamazom bileşenlerinin ve hedef proteinlerin ekspresyonunu arttırır. İkinci "aktivasyon" sinyali inflamazom oluşumunu sağlar. İkinci sinyal, serbest oksijen radikalleri (ROS), lizozomal hasar ve potasyum akışı dahil olmak üzere üç ana mekanizma içerir.
NLRP3 aktivasyonu için iyon akışı modeli, ROS modeli ve lizozomal rüptür modeli olmak üzere 3 ana model öne sürülmüştür. İyon akışı modelinde spesifik katyonlar olan K+, H+, Ca+2 hücre içi seviyelerindeki değişikliklerin NLRP3 inflamazom aktivasyonunu sağladığı öne sürülmektedir. Ekstraselüler ATP, ATP bağmlı iyon kanalı olan PX27’yi aktive ederek hücre dışına K+ çıkışına neden
olmaktadır [38-39]. Nigerisin hücre membranında porlar oluşturarak hücre dışına K+
çıkısını [34], İnfluenza M2 proteini H+ iyonlarının golgiden sitoplazmaya salınımını
tetiklemektedir [40]. Yüksek ekstraselüler Ca+2 konsantrasyonu, hücre içi Ca+2 miktarının artmasına ve CAMP aktivasyonuna neden olmaktadır [41]. Tüm bu hücre içi iyon değişiklikleri NLRP3 inflamazomunu tetikleyerek IL-1β ve IL-18 salınımınına neden olmaktadır. İyon akışının NLRP1b [42-43], NLRC4 gibi diğer
inflamazomları da aktive ettiği ve bu nedenle de sadece NLRP3’e özgü olmadığı görülmüştür [44].
NLRP3 aktivasyonu yapan ATP, ürik asit, nigerisin gibi birçok uyaran ROS üretimini arttırmaktadır [18]. ROS direk olarak NLRP3 aktivasyonunu sağlamamakta, NF-kB üzerinden NLRP3 inflamazomunu aktive ederek pro IL-1β üretimini arttırmakta ve 1. sinyalin oluşumunu sağlamaktadır [45]. Yapılan son çalışmalardan birinde hücre içi artan ROS’ların tioredoksin ve tioredoksin etkileşim protein (TIP) tarafından algılandığı ve bu proteinlerin NLRP3’e bağlanarak inflamatuar yanıtı tetiklediği gösterilmiştir. Serbest oksijen radikalleri nedeniyle disfonksiyone olan mitokondriden salınan oksitlenmiş mitokondriyal DNA’nın da NLRP3’e direk bağlanarak aktive ettiğini gösteren çalışmalar da mevcuttur.
Ürik asit kristalleri, alüminyum, silika, hidroksiapatit ve amiloid-β fagositozu sırasında gerçekleşen lizozomal rüptür sonucu NLRP3 inflamazomu aktive olmaktadır [18]. Lizozomal proteazı inhibe eden katepsin B’nin NLRP3 inflamazom aktivasyonunu da inhibe ettiği ve NLRP3 aktivasyonu üzerine proteolitik etki gösterdiği öne sürülmektedir [46].
- AIM2 inflamazomu: AIM2 inflamazomu kaspazı tanımayı sağlayan adaptör protein ASC, inflamazom bağımlı sitokin salınımını ve piroptozisi sağlayan N-terminal PYD ve çift sarmallı DNA’yı (dsDNA) tanımayı sağlayan C-N-terminal HIN 200 domaininden oluşmaktadır. Bakteriyel (Francisella tularensis ve Listeria monocytogenes) ve viral (sitomegalovirus) dsDNA inflamazomun HIN domainine bağlanarak AIM2 proteinin DNA molekülü etrafında oligomerize olmasını sağlamaktadır [47-49]. AIM2, ASC ve kaspaz-1 birleşerek inflamazomu oluşturmakta ve inflamatuar sitokinlerin üretimi başlatılmaktadır.
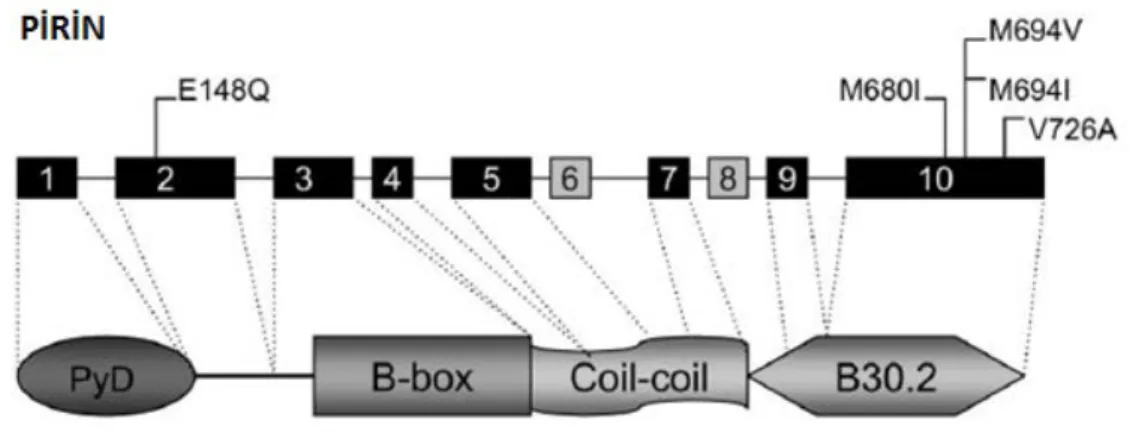
- Pirin inflamazomu: Pirin inflamazomu ASC ile etkileşimi ve kaspaz aktivasyonunu sağlayan bir N-terminal Pirin (PYD) bölgesi, B box çinko parmak bölgesi, bir sarmallanmış sarmal domain (coiled coil) ve bir karboksi ucu B30.2/SRY bölgesinden oluşmaktadır [50]. Hastalıklarla ilişkili mutasyonlar genellikle C-terminal B30.2/SRY bölgesinde görülmektedir. Pirin domaini ASC ile etkileşime girerek inflamazomu oluşturmakta ve böylelikle apoptozis regülasyonunda, inflamasyonda ve IL-1β üretimiminde rol almaktadır [51]. Bir sensör protein de olan pirin Burkholderia
cenocepia gibi Rho GTPaz’ın glikozilasyonu, ADP-ribozilasyonunu ve diğer posttranslasyonel modifikasyonlarını değiştiren bakteriyel toksinleri algılayarak inflamazom oluşumunu ve inflamasyonun başlamasını tetiklemektedir [52-53].
Şekil 2.4. Pirin aksiyon mekanizmaları [54] a-) Sekestrasyon hipotezi : Pirin’in PYD domaini ile
kriyopirinin PYD domaini ASC’nin PYD domainine bağlanmak için yarışır. Pirin –ASC etkileşimi sonucu kriyopirin ASC’ye bağlanamaz bunun sonucunda kaspaz-1 aktivasyonu ve IL-1β üretimi inhibe olur (pirinin inflamasyon oluşumdaki negatif etkisi). b-) Pirin
inflamazom hipotezi: PAMP’lar pirinin C-terminal B30.2 bölgesine bağlanarak inflamazom
oluşumunu tetikler. Bunun sonucunda kaspaz-1 aktivasyonu ve IL-1β üretimi aktive edilir (Pirinin inflamasyon oluşundaki pozitif etkisi).
2.3.2.2. Noncanonical İnflamazom
Noncanonical inflamazom yolakları farelerde kaspaz-11 insanlarda kaspaz 4 ve 5 aktivasyonu ile sonuçlanmaktadır [55-56]. TLR4 bakteryel lipopolisakkaritleri algılar ve kaspaz-11 aktive edilir. Kaspaz- 11 de NLRP3 inflamazomunu aktive ederek proinflamatuar sitokinlerin salınmasını sağlar. İntraselüler lipopolisakkaritler ve bazı bakteriler TLR4’den bağımsız olarak IL-1a sekresyonu ve piropitozisi gerçekleştirirler fakat bu yolla IL-1 β sekresyonu sağlanamamaktadır [57]. Ayrıca insanlardaki kaspaz-4 noncanonical yolağı kullanmadan direk olarak da NLRP3 inflamazomunu aktive edebilmektedir [58].
2.3.2.3. ASC ve İnflamasyonun/inflamazomun yayılımı
ASC sağlıklı kişilerin monosit ve makrofajlarında nükleusta lokalize iken patojenle enfekte olduktan sonra hızlıca sitozole doğru yer değiştirmekte kaspaz-1 ve NOD-benzeri reseptörden oluşan inflamazomun oluşmasını ve piropitozisi sağlamaktadır. ASC’nin nükleustan sitozole geçişi engellendiğinde inflamazom oluşumunun da engellendiği gösterilmiştir [59]. Yapılan son çalışmalarda piropitozis ile ölen hücrelerden salınan ASC’nin ekstraselüler alanda toplanarak pro-IL-1β’nın matur hale gelmesini sağladığı ve kaspaz-1’i aktive ettiği gösterilmiştir. Böylelikle inflamasyonun yayılmasında etkili olduğu gösterilmiştir [60].
İnflamazomlar sensör proteinlerle patojenleri algılayarak inflamasyon yanıtını başlatırlar. Bu sensör proteinleri kodlayan genlerde defekt olması durumunda otoimmun ve otoinflamatuar hastalıklar meydana gelmektedir. Aşağıdaki tabloda (Tablo 2.1) inflamazomların koruduğu enfeksiyöz ajanlar ve inflamazomları oluşturan proteinlerdeki genetik defektler sonucu oluşan hastalıklar gösterilmektedir. Tabloda görüldüğü üzere Kriyopirin ilişkili periyodik sendromlar (CAPS) ve Ailevi Akdeniz Ateşi (FMF) inflamazomopati grubu hastalıklardır.
Tablo 2.1. Hastalıklara karşı inflamazomların koruyucu ve zararlı etkileri [24]
K O R U Y U C U B.anthracis C.rodentium E.coli S.Typhimurium IBD L.pneumophila P.aeruginosa S.typhimurium S.flexneri Y.pestis IBD F.tularensis L.monocytogenes Sitomegalovirus
Çiçek virusu B.cenocepacia
. NLRP1 NLRP3 NLRC4 AIM2 PİRİN Z A R A R L I Addison hast. SLE Tip 1 DM Vitiligo Artrit Çölyak hast. CAPS Schnitzler send SLE Tip 1 DM FCAS- benzeri MAS –benzeri SCAN4 SLE Tip 1 DM FMF
2.4. Ailevi Akdeniz Ateşi (FMF)
Otoinflamatuar hastalıkların en sık görüleni FMF tekrarlayan ateş atakları ve seröz zarların tutulumu ile karakterize bir hastalıktır.
2.4.1. Epidemiyoloji
Akdeniz kökenli etnik gruplarda sık görülmektedir. Sefardik Yahudi, Ermeni, Yunan, Arap ve Türk toplumunda sık görülmektedir. Yapılan çalışmalarda Ermenilerde taşıyıcılık oranı 1/7, hastalık insidansı 1/500 olarak belirtilmektedir [61]. Taşıyıcılık oranın Irak’lılarda 1/4'e kadar yükseldiği belirtilmektedir.
Şekil 2.5. İnfevers veri tabanında MEFV geninde saptanan mutasyonlar 2.4.2. Ailevi Akdeniz Ateşi’nde Genetik
FMF, 16.kromozomun kısa kolunda (16p13.3) lokalize olan ve 10 ekzondan oluşan MEFV gen mutasyonları sonucu meydana gelmektedir [62]. MEFV geni 781 aminoasitten oluşan pirin yada marenostrin olarak adlandırılan bir proteini kodlamaktadır [63-64]. Infevers veri tabanına göre bugüne kadar 310’dan fazla MEFV sekans varyantı bildirilmiştir (Şekil 2.5). Bildirilen tüm varyantlar hastalıkla ilişkili bulunmamıştır. Yeni mutasyonların bulunmasıyla sadece sık görülen mutasyonların bakılmasının yeterli olup olmayacağı tartışmalı hale gelmiştir. Booty ve ark. yaptığı
çalışmalar sonucunda FMF kliniği olan olgularda MEFV gen dizi analizi yerine sık görülen mutasyonların taranmasının tanı için yeterli olacağını öne sürmüştür [65]. 2012 yılında bir grup klinik ve moleküler uzman 14 MEFV gen varyantının bakılması konusunda görüş birliği sağlamıştır. Bu genlerden 9’u kesin patojenik olan (M694, M694I, M680I, V726A, R761H, A744S, I692del, E167D ve T267I mutasyonu) ve 5’i de önemi tam bilinmeyen varyant (E148Q, K695R, P369S, F479L ve I591T mutasyonu) olarak belirtilmiştir [66].
M694V mutasyonu Türk, Ermeni, Arap toplumlarında en sık görülen mutasyon olarak bildirilmiştir [67-70]. İkinci en sık görülen mutasyon da Türk’lerde M680I mutasyonu, Ermeni, Arap ve Yahudi’lerde V726A mutasyonu olarak bildirilmiştir. M694V homozigot mutasyonları daha ağır klinik gidiş ve erken yaşta başlangıçla karakterizedir. Sık görülen mutasyonlardan olan E148Q mutasyonun hastalık yapıcı etkisi ise tartışmalıdır [71]. Ben-Chetrit ve ark. 2000 yılında yaptığı çalışmada sağlıklı populasyonla hasta popülasyonunda E148Q mutasyon sıklığının benzer olduğunu [72], Tchernitchko ve ark. yaptığı çalışmalar sonucu E148Q mutasyonunun benign bir polimorfizm olabileceğini bildirmişlerdir. Yapılan diğer çalışmalar sonucunda da homozigot E148Q mutasyonun FMF - benzeri fenotip oluşturduğu bu nedenle kolşisin tedavisinin başlanması önerilmiştir [73]. E148Q mutasyonunun daha geç yaşta başlangıç, daha hafif seyirli hastalık, ekzon 10 mutasyonlarına göre kolşisine daha iyi yanıt verme ile karakterize olduğu belirtilmiştir [74-75]. Shinar ve ark. E148Q mutasyonu önemi belirsiz varyant olarak değerlendirmiş ve bu nedenle E148Q mutasyonun tek MEFV gen varyantı olması durumunda FMF tanısı konamayacağını öne sürmüştür [76].
FMF otozomal resesif kalıtımlı bir hastalık olmasına rağmen birçok hastada MEFV geninde tek mutasyon olduğu görülmüştür. Marek-Yagel ve ark. yaptığı çalışmada bazı olgularda heterozigot ve homozigot mutasyona sahip olmanın klinik olarak ayırtedilemediği bu nedenle de FMF ‘in otozomal dominant kalıtımlı fakat düşük penetranslı olabileceğini öne sürmüşlerdir [77]. Son zamanlarda yapılan bir çalışmada tek düşük peneteranslı mutasyona sahip olgularda FMF benzeri semptomların daha az sıklıkta görülmesine karşılık 2 yüksek penetranslı mutasyona sahip bireylerde FMF benzeri semptomların daha sık olması nedeniyle mutasyonlarla ilişkili bir ‘doz etkisi’ olabileceğini öne sürmüştür [78]. Çevresel faktörlerinde hastalık fenotipini etkilediği bilinmektedir.
MEFV geninde görülen H478Y, T577S, T577A, T577N, M694del, M694I, E148Q ve L110P mutasyonlarının otozomal dominant kalıtımla FMF’e neden olduğu birçok yayında belirtilmiştir. Otozomal dominant kalıtımla seyreden olgularda hastalığın daha geç başlangıçlı olduğu ve ataklarda diğer otoinflamatuar hastalıklarda görülen ürtikeryal raş ve konjonktivit gibi bulgularında da görülebildiği belirtilmektedir[63][79][80].
Şekil 2.6. MEFV geni ve kodladığı pirin proteini 2.4.3. Patogenez
MEFV geninin kodladığı pirin ASC ile etkileşime girerek inflamazom oluşmasını sağlamaktadır. İnflamazomlar kaspazları aktif hale getirerek IL-1β salınımı tetiklemektedir. Hastalık yapıcı mutasyonların işlev kaybı ya da işlev kazanımı sonucu oluştuğu yakın zamana kadar tartışılmıştır. Fonksiyon kaybı modelini savunan Papin ve ark. yaptıkları çalışmalarda pirin inhibe edildiğinde kaspaz-1 aktivasyonun ve IL-1β sekresyonun arttığını göstermişlerdir [81]. Hesker ve ark. yaptıkları çalışmada MEFV geninden yoksun farelerde inflamatuar uyarılara karşı artmış IL-1β yanıtının olduğunu göstermiştir [82]. Diğer taraftan fonksiyon kazanımı modelini savunan Booty ve ark. FMF hastalarında pirin ekspresyonunun sağlıklı kişilere göre daha fazla olduğunu göstermiştir [65]. Yu ve ark. yaptıkları çalışmada pirin proteinin ASC ve PSTPIP1 ile birleşerek direk olarak kaspaz-1 aktivasyonu yaptığını ve IL-1β sekresyonunu arttırdığını göstermiştir [83]. Bu sonuçlarla FMF mutasyonlarının fonksiyon kazanım mutasyonları sonucunda oluştuğu ve FMF’in bir pirin inflamazomopatisi olduğu doğrulanmıştır [84].
Bir patojen tanıma reseptörü de olan pirin Clostridium difficile, Clostridiım botulinum ve Burkholderia cepacia bakterilerinin RhoGTPaz üzerinde yaptığı modifikasyonları algılayarak kaspaz-1 aktivasyonu ve IL-1β salınımını gerçekleştirmektedir [53][85]. Protein kinazlar RhoA’yı aktive etmektedir. Park ve ark. yaptıkları çalışmada bu protein kinazların (PKN) pirin inflamazomunu suprese ettiğini, B30.2 mutasyonları olan M694V/M694V, M680I/M680I, V726A/V726A mutasyonlu farelerde PKN1’in pirine bağlanmasının azaldığını, B30.2 domainin protein kinaz N1’in (PKN1) pirine bağlanmasını düzenlediğini göstermiştir [86]. Bu veriler ile mutasyonların ve RhoA’nın pirin inflamazomu üzerindeki inhibitör etkisi aydınlatılmıştır.
Pirin nötrofillerde, monositlerde, peritoneal ve sinovial fibroblastlarda eksprese edilmektedir. Pirin nötrofillerin sitoplazmasında ve mikrotubuluslarda yerleşir. Mikrotubuluslar pirinin defosforilasyonu ile seyreden sinyal yolağını kontrol etmektedirler. Van Gorp ve ark. yaptıkları çalışmada mutant pirin inflamazomunun ASC nükleasyonu için mikrotubullere ihtiyaç duymadığını, MEFV genindeki B30.2 mutasyonlarında mikrotubullerden bağımsız olarak pirin aktivasyonu sağladığını göstermiştir [87].
FMF atak sırasında nötrofili görüldüğü ve inflame dokulara nötrofil akışı olduğu bilinmektedir [88]. Gohar ve ark. yaptıkları çalışmada M694V mutasyonuna sahip olguların uyarılmamış nötrofillerinin sağlıklı kişilere göre daha fazla IL-18 ve kaspaz-1 salgıladığını göstermiştir [89]. Diğer bir çalışmada FMF ataklarının, aktif IL-1β da dahil olmak üzere nötrofil hücre dışı tuzakları (NET) salınması ile karakterize olduğu belirtilmiş, NET yapılarının atağın ilk saatlerinden itibaren görüldüğü ve atak sonuna doğru azaldığı gözlenmiştir [88].
2.4.4. Klinik
FMF atakları tekrarlayan ateş ve serozal inflamasyon bulgularıyla (eklem ağrısı, göğüs ağrısı, karın ağrısı) karakterizedir. Olguların çoğunda ataklar erken çocukluk döneminde başlar. %65 olguda atak 10 yaşından önce, %90 olguda 20 yaşından önce başlamaktadır [90]. Ataklar 12 saat ile 3 gün arasında sürmekle birlikte atak aralarındaki süre değişkendir. Genellikle net bir tetikleyici faktör olmamaktadır. Fakat bazı olgularda yoğun egzersiz, soğuğa maruziyet, emosyonel stres, cerrahi işlemler, menstruasyon ve yorgunluğun FMF ataklarını tetiklediği belirtilmektedir.
Tekrarlayan ateş FMF’in en karakteristik özelliklerinden birisidir ve atak sırasında hastaların hemen hemen hepsinde mevcuttur [91]. Ateş 38-40 C arasında 15
seyretmekte ve 12 saat ile 3 gün arasında devam etmektedir. Erken çocukluk döneminde tekrarlayan ateş FMF’in tek semptomu olabilmektedir [92]. Kolşisin tedavisi başladıktan sonra ateşsiz FMF atakları görülebilmektedir.
Karın ağrısı hastaların yaklaşık %95’inde ataklara eşlik etmektedir [90]. Karın ağrısı ve hassasiyet başlangıçta lokalize olabilirken sonrasında jeneralize hale gelmektedir. Karın ağrısının periton inflamasyonuna bağlı olması nedeniyle periton irritasyon belirtileri olan rebaund, defans ve adinamik ileus görülebilmekte ve bu bulgular nedeniyle FMF atakları akut batın tablosu ile karışıp, cerrahi işlemlerin yapılmasına neden olabilmektedir.
Göğüs ağrısı etnik kökene bağlı olmakla birlikte %33-88 olguda görülmektedir [91]. Göğüs ağrısı plevra inflamasyonuna ya da subdiafragmatik inflamasyona bağlı görülmektedir. Göğüs ağrısı genellikle tek taraflı olup nefes almakla ve ve öksürmekle artan ağrı ile karakterizedir. Küçük, geçici plevral efüzyon eşlik edebilmektedir. Ağrı genellikle 3 gün içinde gerilese de 1 haftaya kadar da uzayabilmektedir. Plevritli hastalarda eşlik eden perikardit de görülebilmektedir.
Eklem ağrısı Sefardik Yahudi FMF hastalarında yaklaşık %75 oranında görülmektedir. Minör travma ya da uzun yürüyüş eklem ağrısını tetikleyebilmektedir. Eklem ağrısı genellikle monoartikülerdir ve büyük eklemleri (diz, ayak bileği, kalça) tutmaktadır. Migratuar poliartritli olgularda bildirilmiştir. Sıklıkla sinovyal efüzyon gelişir ve sinovyal sıvı sterildir. 24-48 saat içinde bulgular gerilemekte ve sıklıkla da eklemde deformiteye yol açmamaktadır. Atakları sık tekrarlayan, uzun süren olgularda kalıcı deformite, fonksiyonel snırlanma, etkilenen eklem çevresinde osteoporoz ve aseptik nekroz göülebilmektedir [93].
Erizipel benzeri deri lezyonları %12-40 FMF olgusunda görülmektedir [94]. Bu ağrılı, hassas, deriden kabarık ve kızarık lezyonlar daha çok alt bacak, ayak bileği ve ayak üzerinde oluşmaktadır. Bu lezyonlar kendiliğinden gerilemekte ve antibiyotik tedavisi gerektirmemektedir. Atak dönemlerinde miyalji ve erizipel benzeri deri lezyonları olan olgularda ataklar arası dönemde subklinik inflamasyonun devam ettiği bu dönemlerde akut faz reaktanlarının yüksekliği gösterilerek kanıtlanmıştır [95].
Daha nadir görülen semptomlardan biri olan egzersiz myaljisi daha çok alt ekstremitede ağrıya neden olmaktadır. Epizodik karakterde değildir ve kolşisin tedavisine de yanıt vermez. Dinlenme ya da non-steroid antiinflamatuar kullanımı ile yakınmalar gerilemektedir.
Semptomatik akut perikardit FMF hastaların < %1’den görülmektedir [96]. Yapılan bir çalışmada perikardit sıklığının FMF hastalarında sağlıklı popülasyona göre 11 kat artmış olduğu gösterilmiştir [97]. Göğüs ağrısı olması, fizik bakıda perikardiyal sürtünme sesi duyulması ve EKG’ de ST segment elevasyonu görülmesi perikarditi desteklemektedir.
FMF’li olgularda skrotumun tek tarafında şişme ile karakterize akut skrotum olarak adlandrılan tablo görülebilmektedir. Testis ve epididimis normal, tunika vajinalis kalın ve hiperemik görünümdedir. Testiküler torsiyondan farklı olan bu tablo cerrahi işlem gerektirmemektedir.
FMF’li olgularda sekiz haftaya kadar uzayabilen ve daha çok alt ekstremiteleri tutan ateşli myalji atakları görülebilir. Bu hastaların eritrosit sedimentasyon (ESR) değerleri artmış olmakla birlikte kreatin kinaz (CK) değerleri ve elektromyogramları (EMG) normaldir. Bu ataklar M694V homozigot mutasyonu olan olgularda görülmesi nedeniyle hastalık ağırlığı ile ilişkili olduğu düşünülmektedir. Etyolojisi belli olmamakla birlikte vaskulitik bir durum olabileceği düşünülmektedir. Tedavisinde glukokortikoidler kullanılmaktadır.
Baş ağrısı FMF ataklarına eşlik edebilmektedir ve sıklıkla hafif seyirlidir. Ciddi baş ağrısı olan ve rekürren aseptik menenjit tablosu olan olgular bildirilmiştir fakat bu tablo oldukça nadir görülmektedir.
Henoch-Schönlein purpurası, poliarteritis nodosa, Behçet hastalığı gibi sistemik non-granülomatöz vaskulitlerin FMF hastalarında görülme insidansının arttığı bbildirilmiştir [98]. Bu olgular sıklıkla M694V homozigot mutasyonuna sahip olgulardır.
2.4.5. Uzun Dönem Komplikasyonları
Sekonder (AA) amiloidozis, FMF hastalarındaki mortalitenin önemli bir nedenidir [99]. Amiloidozis FMF’li bazı olgularda ilk ve tek bulgu olarak görülebilmektedir [100]. Hastalar asemptomatik proteinüriyle görülebileceği gibi nefrotik sendrom, progresif nefropati ve son dönem böbrek yetmezliği ile de karşımıza çıkabilmektedir. Son dönem böbrek yetmezliği proteinürinin başlangıcından 2-13 yıl sonra gelişmektedir. Amiloid birikimi dalak, karaciğer, gastrointestinal sistem, kalp, tiroid ve testiste görülmektedir.
Amiloidoz birikiminin FMF ataklarının sıklığına ya da ağırlığına mı bağlı yoksa genetik defektten mi kaynaklı olduğu hala tartışmalıdır. Kolşisin kullanımı ile 17
amiloidoz insidansında azalma olmuştur [101][67]. Amiloidoz risk faktörleri arasında etnik köken ve ailede amiloidoz öyksü olması da yer almaktadır [102]. M694V ve V726A mutasyonlarının fenotipik ekspresyonuna bakıldığında diğer mutasyonlara göre daha fazla amiloidoz yaptığı da görülmüştür [103-104].
2.4.6. Laboratuvar
Atak dönemlerinde lökositoz, akut faz reaktanlarının (ESR, C-Reaktif protein, serum amiloid A, ferritin) yüksekliği görülmektedir. Ataklar arası dönemde de akut faz reaktanları normal seyretmektedir. Böbrek fonksiyon testlerinde yükseklik ve proteinüri amiloidoz açısından uyarıcıdır.
2.4.7. Tanı
FMF tanısı klinik bulgular ve MEFV geninin moleküler analizi yapılarak genetik konformasyonla konulmaktadır [105]. FMF için farklı sınıflandırma ve tanı kriterleri mevcuttur. İlk oluşturulan tanı kriterleri Tel Hashomer kriterleridir [90] (Tablo2). 1997 yılında Livneh ve ark. Tel Hashomer kriterlerindeki amiloidoz gibi belirtileri çıkararak yeni tanı kriterleri oluşturmuşlardır [106] (Tablo2). Bu kriterler daha çok erişkin için hastalar için yapılmıştır. Çocuk hastalarda atakların daha kısa süreli olması, çoğu olguda tek taraflı göğüs ağrısının olmaması, atakların daha çok tekrarlayan ateşle ya da sadece ateşle seyretmesi, bazı pediatrik hastaların ağrının yerini ve şiddetini tam olarak ifade edememesi gibi nedenlerle pediatrik FMF kliniğini erişkin hastalardan farklı seyretmektedir. Bu nedenle Tel Hashomer kriterlerinin pediatrik hastalar için çok uygun olmadığı düşünülerek pediatrik hastalar için yeni tanı kriterleri oluşturulmuştur [107] (Tablo2). Türk FMF pediatri kriterleri denen bu kriterlerin 5’inden 2’sinin olması durumunda sensitivitesi %88, spesifitesi %92.2 olarak belirtilmiştir [107].
2.4.7.1. Genetik Tanı
2012 yılında FMF de dahil olmak üzere herediter periyodik ateş sendromlarının genetik tanı testleri için bir kılavuz yayınlanmıştır [66]. 9’u patojen (M694V, M694I, M680I, V726A, R761H, A744S, E167D, T267I, I692del) diğer 5’i 18
patojenitesi net olmayan (K695R, E148Q, P369S, F479L, I591T) bu mutasyonların taranması önerilmiştir.
Tablo 2.2. FMF tanı kriterleri (Tel Hashomer, Livneh, Türk Pediatrik FMF kriterleri) Tel Hashomer kriterleri
Majör kriterler:
1.Peritonit, plörit veya sinovitin eşlik ettiği tekrarlayan ateşli epizodlar 2.Predispozan bir hastalık olmaksızın AA-tip amiloidoz
3.Devamlı kolşisin tedavisine anlamlı yanıt
Minör kriterler:
1.Tekrarlayan ateşli epizodlar 2.Erizipel benzeri eritem
3.Birinci derece akrabalarda AAA öyküsü Kesin tanı: 2 majör veya 1 majör ve 2 minör kriter
Livneh FMF tanı kriterleri Majör kriterler (tipik atak)
1.Peritonit (yaygın)
2.Plevrit (tek taraflı) ya da perikardit 3.Monoartrit (kalça, diz, ayak bileği) 4.Tek başına ateş
5.İnkomplet abdominal atak
Minör kriterler
Aşağıdaki bölgelerden herhangi birini ya da ikisini etkileyen inkomplet ataklar
1.Göğüs 2.Eklem
3.Yorgunluk sonrası bacak ağrısı 4.Kolşisine iyi yanıt
Destekleyici kriterler
1.Aile öyküsü 2.Uygun etnik köken
3.Hastalık başlangıç yaşının <20 olması Atağın özellikleri:
4.Yatak istirahati gerektirecek kadar ağır olması 5.Kendiliğinden düzelmesi
6.Belirtisiz dönemler olması
7.Beyaz küre sayımı, ESR, SAA, fibrinojen testlerinden herhangi birinde ya da birkaçında yükseklik olması
8.Epizodik proteinüri/hematüri
9.Patolojik olarak doğrulanmayan apandisit öyküsü 10.Anne baba arasında akrabalık olması
Tanı: 1 major kriter / 2 minör kriter /1 minör + 5 destekleyici kriter / 1 minor + ilk 5 destekleyici kriterden 4’ü
Pediatrik Türk FMF kriterleri
1.Ateş (aksiller >38 C, 6-72 saat süren 3 veya daha fazla atak) 2.Karın ağrısı (6-72 saat süren 3 veya daha fazla atak) 3.Göğüs ağrısı (6-72 saat süren 3 veya daha fazla atak) 4.Artrit (oligoartrit, 6-72 saat süren 3 veya daha fazla atak) 5.FMF aile öyküsü
Tanı: 5 kriterden 2’si olması yeterlidir.
2014 yılında da FMF’in genetik tanısı için yayınlanan kanıta dayalı güncel tavsiyeler şunlardır [76];
1-FMF genetik test ile desteklenebilecek ancak dışlanamayacak bir klinik tanıdır. 19
2-M694V homozigot hastalar daha ağır klinik seyir gösterme riskini taşıdıkları göz önüne alınmalıdır.
3-Homozigot ya da birleşik heterozigot mutasyon (özellikle M694V ya da ekzon 10’daki 680-694 pozisyonundaki mutasyonlar) taşıyan FMF hastalarının kliniği daha ağır seyredebileceği bilinmelidir.
4-Patojenik özelliği net olmayan E148Q mutasyonunun tek başına varlığı FMF tanısını desteklememektedir.
5-M694V homozigot mutasyonu olan hastalar erken başlangıçlı hastalık için risk altındadır.
6-M694V homozigot olan ve semptomu olmayan olgular değerlendirilmeli ve tedavi için yakın takip edilmelidir.
7-FMF için 2 patojenik mutasyona sahip, semptomu olmayan fakat amiloidozis için risk faktörleri (etnik köken, aile öyküsü, persistan akut faz reaktan yüksekliği) olan bireyler yakın takip edilmeli ve tedavi başlanması düşünülmelidir.
8-Bir otoinflamatuar hastalıklar uzmanı ile konsültasyon tanı, genetik test endikasyonu ve yorumu için yardımcı olabilir.
2.4.7.2. Ayırıcı tanı
FMF’in ayırıcı tanısında diğer periyodik ateş sendromları, sistemik juvenil idiopatik artrit, vaskulitler, enfeksiyonlar, maligniteler ve akut batın tablosuna yol açan hastalıklar yer almaktadır.
2.4.8. Tedavi
1972 yılından itibaren kullanılmaya başlayan kolşisin halen FMF’in ana tedavisini oluşturmaktadır [108]. Kolşisin atak sıklığını ve şiddetini azaltmakta hastaların hayat kalitesini yükseltmektedir [109]. Livneh ve ark. yaptıkları çalışma kolşisinin FMF hastalarında sekonder amiloidoz oluşumunu da engellediğini göstermiştir [110].
Pediatrik FMF hastalarında infantlarda dahil kolşisin iyi tolere edilmektedir [111]. En sık görülen yan etkileri ishal, kusma ve karaciğer fonksiyon testlerinde yükselmedir. Bu etkiler %5-10 olguda tavsiye edilen doz aralığında kolşisin tedavisi almasına rağmen görülebilmektedir.
Kolşisin FMF ‘in ana tedavisi olmakla birlikte olguların 1/3’ünün kolşisin tedavisi ile parsiyel remisyonda olduğu, %5 kadarının kolşisine yanıtsız olduğu, %2-5 olgunun da gastrointestinal sistem yan etkileri nedeniyle kolşisini tolere edemediği bilinmektedir [112]. Lidar ve ark. yaptıkları çalışma sonucunda kolşisin tedavisine yanıtta hastanın sahip olduğu MEFV gen mutasyonun önemli olduğunu göstermiştir [113]. M694V homozigot mutasyonu olan olgularda klinik daha ağır seyretmekte ve yüksek doz kolşisin ihtiyacı olmaktadır.
Son yıllarda kolşisine yanıtsız olgularda biyolojik ajan kullanımının faydalı olduğunu gösteren birçok çalışma yayınlaşmıştır [114]. Pirinin IL-1 aktivasyonunu regüle edici etkisi üzerinden anti-IL-1 tedavilerinin kolşisine dirençli FMF olgularında inflamasyonu baskılamada başarılı olduğu gösterilmiştir. IL-1 reseptör antagonisti olan anakinra, IL-1 resptörüne bağlanmak için IL-1α ve IL-1β ile yarışmaktadır. İnsan dimerik füzyon proteini olan rilonacept, IL-1α ve IL-1β’nın ekstraselüler domainine bağlanmaktadır [115]. Canakinumab da direk IL-1β ya karşı olan insan IgG1 monoklonal antikorudur [115]. Anakinra tedavisi subkutan olarak her gün, rilonacept tedavisi subkutan olarak haftada bir, canakinumab tedavisi subkutan olarak ayda bir uygulanmaktadır. Kolşisine dirençli 14 FMF hastasında Rilonacept ile yapılan randomize kontrollü bir çalışmada FMF ataklarının azaldığı fakat atak süresinde değişme olmadığı görülmüştür [116]. Canakinumab ile ilgili yapılan çalışmalarda kolşisine yanıtsız olgularda canakinumab tedavisinin atak sıklığını ve şiddetini azalttığı gösterilmiştir. Fakat bu çalışmalardaki olgu sayısının azlığı nedeniyle daha geniş ve kontrollü çalışmaların yapılması gerekmektedir.
Yakın dönemde İsrailli ve Fransız uzmanlar tarafından FMF tedavisinin yönetimi için kanıta dayalı tavsiyelerde bulunmuşlardır [109]. Bu tavsiyeler kolşisin dozu, çocuk ve erişkinlerde maksimum kolşisin dozu, kolşisin direncinin tanımı ve kolşisine dirençli olgularda alternatif tedaviler ile ilgilidir. Bu tavsiyeler şunlardır;
• Takip eden 3 ayda her ay birden fazla atağı olan ya da akut faz reaktanları sürekli yüksek seyreden olgularda kolşisin dozu arttırılmalıdır
• Yılda altıdan fazla tipik FMF atağı geçiren olgular kolşisin dirençli olarak kabul edilmelidir.
• Maksimum kolşisin tedavisine rağmen (çocuklarda 2 mg/gün) FMF atakları sık tekrarlıyorsa uzmanlar IL-1 inhibitör tedavilerini önermelidir.
Etanercept, infliximab, adalimumab gibi TNF blokerlerin kronik artriti yada sakroileiti olan FMF hastalarında kullanımının faydalı olabileceğini gösteren çalışmalar da mevcuttur [117].
2.5. Tümör Nekroz Faktör Reseptör 1 İlişkili Periyodik Sendrom (TRAPS)
TNF-reseptör 1 ilişkili periyodik sendrom, nadir görülen, otozomal dominant kaltımlı bir multisistem otoinflamatuar bir hastalıktır.
2.5.1. Epidemiyoloji
İlk kez 1982 yılında İskoç kökenli geniş bir ailede tanımlanması nedeniyle ‘Ailesel Hibernian Ateş’ olarak adlandırılmıştır. İlerleyen dönemlerde Amerikan, Japon ve Akdeniz kökenli başka olgularda bildirilmiştir. Tekrarlayan ateş, myalji, raş, karın ağrısı ile karakterize olan bu hastalığın prevalansı yaklaşık 1/1.000.000 olarak bildirilmiştir [118].
Şekil 2.7. İnfevers veritabanına göre TNFRSF1A geninde saptanan mutasyonlar
2.5.2. Tümör Nekroz Faktör İlişkili Periyoidk Sendrom’da Genetik
TRAPS 12. kromozomun kısa kolunda (12p13) yer alan TNFRSF1A genindeki mutasyona bağlı gelişir [119]. Bu gen 55-kD TNF alfa reseptörünü (TNFRSF1A) kodlamaktadır. 10 ekzonlu olan TNFRSF1A genindeki mutasyonlar ekzon 2, 3, 4 ve 6 ekzonlarda yoğunlaşmıştır. Mutasyonların lokalizasyonuna göre yüksek ya da düşük penetranslı oldukları ayırt edilmektedir. Mutasyonlar sıklıkla tek nükleotid missense (kayıp) mutasyonları şeklinde görülmekte ve reseptörün 3 boyutlu yapısı için temel bölüm olan ilk iki N-terminal sistinden zengin bölgeler CRD1 ve CRD2 bölümlerinde görülmektedir [120]. Sistein rezidülerinin olduğu bölgede mutasyonları olan bireyler daha ağır klinik seyir göstermekte ve daha yüksek amilodoz riski taşımaktadır [121]. Düşük penetranslı mutasyonlar olan R92Q ve P46L mutasyonları düşük amiloidoz riski, daha geç yaşta başlangıç, hafif yada atipik klinik prezentasyonla seyretmektedir [122].
Şekil 2.8. TNFRSF1A geni ve kodladığı TNF alfa reseptör proteini
TNFRSF1A geninin sağındaki dört numaralı yarı daire, TNF reseptörünün dört hücre dışı sisteinden zengin alanını (CRD 1-4) göstermektedir. Gri zar ile gösterdiği transmembran bölge ve intraselüler ölüm alanı (DD), sinyalleme NF-kB aktivasyonu veya apopitoz gibi olaylar başlatılmasını sağlar [123].
2.5.3. Patogenez
TNF alfa ve TNF reseptör ailesi lokal ve sistemik inflamasyonun tetiklenmesinde anahtar rol oynamaktadır [124]. Tip 2 transmembran proteini olan TNF alfa apopitoz, hücre proliferasyonu, immun modülasyon, otoimmun hastalıklar da dahil olmak üzere birçok biyolojik sürece aracılık etmektedir [125]. Monosit, makrofaj, mast hücresi, T ve B lenfosit, natural killer, nötrofil, eozinofil, düz kas, kalp kası, fibroblast ve osteoklastlarda sentezlenmektedir [126-127]. Bilinen tüm TNF fonksiyonları TNFR1 ve TNFR2 olarak adlandırılan ve NF-kB, mitojen aktive edici protein kinaz (MAPK) gibi sinyal yolaklarını aktive eden iki reseptörden birine bağlanılmasıyla gerçekleşir [128]. TNFR1, 4 adet sisteinden zengin tekrarlar içeren bir ekstraselüler bölge, bir transmembran bölgesi, bir intraselüler ölüm bölgesi (DD) içermektedir [129]. Pre-ligand montaj alanı (PLAID) olarak da adlandırılan N-terminal sisteinden zengin alan CRD1, ligand bağlama ve sinyal iletimine izin verir [130]. Ekstraselüler bölge TNF alfa’nın bağlanma bölgesini oluşturmakta ve proteinin kendi kendine toplanmasını sağlamaktadır.
TNF alfa TNFR1 reseptörüne bağlanır. TNF reseptörünün ölüm bölgesi (DD), TNFR ilişkili ölüm bölgesi proteini (TRADD), TNF reseptör ilişkili faktör (TRAF), reseptör etkileşim proteinin (RIP) oluşturduğu makromolekül ile etkileşime girerek NF-kB sinyal yolağını aktive etmektedir. NF-kB yolağının aktivasyonu ile sitokin üretimi ve bir prokaspaz-8 benzeri apopitoz regülatörü olan FLICE-benzeri inhibitör protein (FLIP) üretimi tetiklenmektedir. FLIP kaspaz 8’i aktive etmekte ve apopitoz tetiklenmektedir. Aktivasyon sonrası TNFR1 reseptörünün ekstraselüler parçası metalloproteinazlar tarafından bölünerek serbest hale getirilir. Bu solubl TNF reseptörleri TNF alfa’ya bağlanarak inflamasyonu durdurur.
TRAPS patogenezi ile ilgili birçok çalışma yapılmıştır. Başlangıçta, TRAPS hastalarının yapısal olarak aktif veya aşırı duyarlı bir TNFRSF1A eksprese ettiği, NF-κB'nin aşırı aktivasyonu ve proinflamatuar sitokin üretiminin artmasıyla hastalığın oluştuğu hipotezi ileri sürülmüştür. Yapılan transfeksiyon çalışmalarıyla hem mutant hem de vahşi tip TNFR1'in NF-κB'yi eşit olarak aktive ettiği bulunmuştur [131]. Daha sonra C33Y, T50M, C52F ve C88R de dahil olmak üzere yapısal mutasyonların protein yapısında değişikliğe yol açarak işlevsel değişikliğe ve membrana bağlı TNFR1'in bozulmuş bölünmesine neden olabileceğini ve bunun da inflamasyon kontrol kaybına neden olabileceğini belirten ‘dökülme hipotezini’ öne sürülmüştür. 24
![Şekil 2.3. NLRP3 inflamazom aktivasyonu için iki aşamalı sinyal [37]. NLRP3, ASC ve pro-kaspaz 1’den oluşan NLRP3 inflamazomunun aktivasyonu, iki aşamalı sinyallerle sıkı bir şekilde düzenlenir](https://thumb-eu.123doks.com/thumbv2/9libnet/3035780.2612/21.892.218.764.202.604/inflamazom-aktivasyonu-asamali-inflamazomunun-aktivasyonu-asamali-sinyallerle-duzenlenir.webp)
![Şekil 2.4. Pirin aksiyon mekanizmaları [54] a-) Sekestrasyon hipotezi : Pirin’in PYD domaini ile](https://thumb-eu.123doks.com/thumbv2/9libnet/3035780.2612/23.892.172.782.237.632/sekil-pirin-aksiyon-mekanizmalari-sekestrasyon-hipotezi-pirin-domaini.webp)
![Tablo 2.1. Hastalıklara karşı inflamazomların koruyucu ve zararlı etkileri [24]](https://thumb-eu.123doks.com/thumbv2/9libnet/3035780.2612/24.892.169.786.701.1003/tablo-hastaliklara-karsi-inflamazomlarin-koruyucu-zararli-etkileri.webp)