T.C.
EGE ÜNİVERSİTESİ TIP FAKÜLTESİ
ÇOCUK SAĞLIĞI VE HASTALIKLARI ANA BİLİM DALI PROF. DR. SAVAŞ KANSOY
GAUCHER HASTALIĞI İZLEMİNDE UZUN SÜRELİ
SONUÇLAR
UZMANLIK TEZİ
Dr. Elvin ORUJOV
DANIŞMAN
Prof. Dr. Mahmut ÇOKER
ÖNSÖZ
Çocuk sağlığı ve Hastalıkları uzmanlık eğitimin süresince, bilgi ve deneyimleri ile eğitimime katkıda bulunan ve kendimizi bu büyük ailenin bir parçası gibi hissettiren, saygıdeğer hocamız, Çocuk Sağlığı ve Hastalıkları Anabilim Dalı Başkanı Sayın Prof. Dr. Savaş KANSOY’a,
Uzmanlık eğitimim sırasında bilgi ve deneyimlerini aktararak yetişmemde büyük emekleri olan, daima yardım ve desteklerini gördüğüm, tez konumun seçimi ve yürütülmesinde katkılarını esirgemeyen değerli hocam Metabolizma – Beslenme Bilim Dalı BaşkanıSayın Prof. Dr. Mahmut Çoker’e,
Asistanlık sürecim boyunca Ege Üniversitesi Çocuk Hastanesi’nde bana mesleğimi öğreten, çok değerli ÇOCUK SAĞLIĞI VE HASTALIKLARI ÖĞRETİM ÜYELERİNE,
Tezimin oluşturulmasında büyük katkı sağlayan ve desteklerini esirgemeyen çok değerli Metabolizma – Beslenme Bilim Dalı Öğretim Üyelerine, Uzman Doktorlarına ve Poliklinik çalışanlarına, özellikle Uzm. Dr. Ebru Erbaş Canda, Uzm. Dr. Havva Yazıcı’ya
Uzmanlık eğitimim boyunca ve tezimin olgunlaşmasında hep yanımda olan ve desteğini esirgemeyen değerli abim Uzm. Dr. Fırat Ergin’e
Tezimin olgunlaşması sırasında kendi uzmanlık alanında soru ve isteklerimi geri çevirmeyen Uzm. Dr. İlker Özgür Koska ve bioistatistik Ar. Gör. Semiha Özgül’e
Uzmanlık eğitimim boyunca hep yanımda olan, sevgisi ve desteğiyle daima bana güç veren sevgili eşim Shola Orujova’ya,
Hayatımın ilk gününden bu yana sevgileriyle her zaman ve her koşulda arkamda olan, sahip olduğum her başarıyı onlara borç bildiğim sevgili annem Niyar Najafova, sevgili babam Arzuman Orujov, sevgili kardeşlerim Mansuma Aliyeva ve Gülnar Orujova’ya,
Bu dört yıllık zorlu süreçte beraber çalıştığım tüm eşkıdemlilerime ve asistan arkadaşlarıma
ÖZET
Gaucher hastalığı (GH), beta glukoserobrozidaz enzim eksikliği ile karakterize, lizozomal lipit depo hastalığıdır. Otozomal resesif kalıtımla geçer. Glukoserebrozidaz enzimini kodlayan genin mutasyonu sonucunda makrofajlarda glukoserebrosid depolanması söz konusudur. Nörolojik tutulum ve nörolojik hastalığa ilerleme durumuna göre 3 alt grubu vardır: Tip 1 (kronik nonnöropatik tip), tip 2 (akut nöronopatik veya infantil tip), tip 3 (kronik nöronopatik veya jüvenil tip).
Ege Üniversitesi Çocuk Metabolizma ve Beslenme Bilim Dalında Gaucher hastalığı nedeniyle izlenen hastalar klinik, laboratuvar bulguları, aldıkları tedavi ve yanıtları, uzun dönem izlem sonuçları değerlendirilmesi hedeflendi.
Çalışmaya toplam 42 hasta dahil edildi. Hastaların 34 tanesi (% 81) tip1, 7 tanesi (% 16,7) tip 2 ve 1 tanesi tip 3 tanılı idi. Ortalama yaş 25,5 yıl (8 ay – 65 yıl), ortalama tanı yaşı 12 yıl (2 ay – 55 yıl) bulundu. En sık başvuru yakınması karın şişliği ve sitopeni idi. Tüm hastalarda tanı enzimatik analiz ve GBA gen analizi ile konuldu. Hastaların 19 tanesinde (% 45,2) anemi, 18 tanesinde (% 42,8) trombositopeni saptandı. Başvuru sırasında 33 hastada (% 80) organomegali, 10 hastada (% 24) büyüme geriliği, 5 hasta (% 12) akut kemik krizleri olduğu görüldü. Sekiz hastada (%19) kemik dansitometre incelemesinde osteoporoz saptandı. Altı hastada (% 14,3) splenektomi öyküsü mevcuttu. Splenektomi olan hastalarda kemik dansitometre Z skor değerleri femur boynu ve vertebra için anlamlı şekilde daha ağır osteoporoz bulgularını göstermekteydi. Bir hasta çocukluk döneminde Gaucher Tip 1 tanısı almsına karşın tedvi edilmemsi nedeniyle Parkinson bulguları gelişmesi sonrası başvurudu. Bir hastada ağır karaciğer yetmezliği gelişti ve karaciğer nakli oldu. Bir tip 3 tanılı kardiyak tutulumu olan hasta kardiyak arrset sonrası eksitus oldu.
İlk tedavi seçeneği tüm hastalar için ERT idi. ERT başlama yaşı 2 ay - 65 yaş
arasındaydı. ERT alma süresi 1 ayla 15 yıl arasında değişmekteydi. ERT tedavi dozu 30 IU / kg ile 120 IU / kg arasında değişmekte idi. Yüksek doz ERT tedavi yanıtına,
hastalığın tipine göre belirlendi. Tip 3 tanılı 5 hasta 60 U/kg`dan, 2 hasta 120U/kg`dan ERT almakta idi. Altı hastada ERT sonrası izlemde SRT tedavisi uygulandı. Tedavi alan hastalarda hemotoljik parametreler, organ volümleri ve kemik bulgularında tedavi sırasında istatistiksel olarak anlamlı düzelme gözlendi. Takip göstergelerinde düşüş saptandı.
Tedaviye ara veren 8 hastada Hb, Hct değerlerinde azalma olduğunu gözlendi, PLT değerinde ise farklılık saptamadı. Dört hastada (% 50) tedaviye ara verdikten sonra göstergelerde yükselme saptandı, 1 hastada organ volümünde artma olduğu gözlendi.
Tedavi sırasında 6 hasta eksitus oldu. Bunlardan 3 hasta tip 1, 1 hasta tip 2, 2 hasta tip 3 tanılıydı.
Gauıcher Tip 1 Hastalığı sık görülen lizozomal depo hastalığıdır. Hemotolojik ve visseral bulgular nedeniyle pek çok hastalıkla benzerlik göstermektedir bu nednele tanı gecikmesi görülebilmektedir. ERT gibi etklin tedavi şeklinin olması nedeniyle erken tanı ve erken tedavi mortalite ve morbidite üzerinde önemli etkiye sahiptir. Bu çalışmada tedavi uygulanan hastalarda tutulan sistemler açısından ERT ve veya SRT uygulaması ile başarılı sonuçlar elde edildiği görülmektedir. Hastaların düzenli takibi, multidisipliner şekilde izlemi sonucu hastaların tanı alması ve etkin tedavi edeilebilmeleri açısından önemlidir.
ABSTRACT
Gaucher’s Disease (GD) is a lysosomal lipid storage disease, which is characterized by beta glucocerebrosidase enzyme deficiency. It has an autosomal recessive genetic nature. There is an accumulation of glucocerebroside due to a mutation in the gene which codes the glucocerebrosidase enzyme in macrophages. This disease is divided into three subtypes according to their neurological involvement and progress into neurological disease as Type 1 (chronic non-neuropathic type), Type 2 (acute non-neuropathic or infantile type and Type 3 (chronic neuropathic or juvenile type).
The main objective was to assess the clinical outcomes, lab results, received treatments and their response and long-term follow-up results of Gaucher’s Disease patients, which are followed by Ege University Pediatric Metabolism and Nutrition Department.
42 patients in total were included in the study. 31(81%) of these patients was diagnosed with Type 1, 7 (16.7%) with Type 2 and 1 was diagnosed with Type 3 of the disease. Mean age was 25,5 and mean age at the initial diagnosis was 12. The most common complaints were abdominal bloating and cytopenia. Anemia was seen in 45.2% of the patients whereas thrombocytopenia was seen in 42.8% of the patients. At the time of admission, 33 (80%) patients had hepatosplenomegaly, 10 (24%) had retarded growth and 5 (12%) had acute bone crises. 8 (19%) patients were diagnosed with osteoporosis in bone densitometry studies. 6 (14.3%) patients had previous splenectomy history. In patients with splenectomy history, Z-score in bone densitometry showed significantly more severe osteoporosis in femur neck and vertebra. One patient presented to our clinic because of developing Parkinson-like symptoms, due to not receiving treatment despite the diagnosis of Gaucher Type 1 in his early childhood. This patient developed severe liver failure and received a liver transplant. One patient diagnosed with Type 3 had cardiac involvement and that patient died due to a cardiac arrest. Complete enzymatic assay and GBA gene analysis was used in all patients for the definite diagnosis.
ERT was the first line of choice for the treatment of all patients. ERT initiation age varied from 2 months to 65 years of age. ERT treatment period
changed from 1 month to 15 years. ERT treatment dosages were between 30 IU/kg and 120 IU/kg. High dosage ERT treatment responses were assessed according to the disease subtype. 5 patients with Type 3 subtype received 60 IU/kg and 2 patients received 120 IU/kg as ERT. SRT was used in 6 patients during post-ERT period. A significant improvement was seen in hematological parameters, organ volumes and bone findings in the patients who received the treatment and their follow-up control values showed a decrease.
8 patients who paused treatment showed a decrease in Hb and Hct values. No difference was detected in platelet levels. 4 patients (50%) showed an increase in their follow-up values after pausing the treatment and increased organ volume was detected in 1 patient.
During treatment period, 6 patients were lost. 3 of those patients had Type 1, 1 patient had Type 2 and 2 patients had Type 3 diagnosis.
İÇİNDEKİLER ÖNSÖZ ... ii ÖZET... iii ABSTRACT ... v TABLOLAR DİZİNİ ... x ŞEKİLLER DİZİNİ ...xii KISALTMALAR ... xiv 1. GİRİŞ VE AMAÇ... 1 2. GENEL BİLGİLER ... 3 2.1. Tanım ve Tarihçe ... 3 2.2. Epidemiyoloji ve Genetik ... 4 2.3. Patofizyoloji ... 5
2.4. Tip 1 Gaucher Hastalığı ... 6
2.5. Tip 3 Gaucher Hastalığı ... 9
2.6. Tip 2 Gaucher Hastalığı ... 11
2.7. Tanı ... 12
2.8. Hastalığın Etkilediği Doku ve Organ Sistemleri ... 13
2.8.1. Hepatosplenomegali ... 13 2.8.2. Hematolojik Değişiklikler ... 15 2.8.3. KemikTutulumu ... 16 2.8.4 Akciğer Tutulumu ... 20 2.8.5. Nörolojik Tutulum ... 22 2.8.6. Hiperimmünoglobulinemi ve Maligniteler... 23 2.9. Tedavi ... 24 2.9.1 ERT ... 24
2.9.1.1. Enzim Replasman Tedavisinin Hedefleri ... 27
2.9.2. Splenektomi ... 28
2.9.3. Kemik İliği Transplantasyonu ... 29
2.9.4. Substrat Azaltıcı Tedavi (SRT) ... 29
2.9.5. Gen Tedavisi ... 30
2.10. İzlem ... 31
2.10.1. Nöronopatik GH`da İzlem ... 32
3. HASTALAR VE YÖNTEM ... 34
3.1. Hastalar ... 34
3.2. Tedavi Protokolü ... 37
3.3. Labaratuar Analizler ... 37
3.3.1. Kuru kan örneğinde Beta Glikozidaz Aktivitesi Tayini ... 37
3.3.2. Glukoserebrosidaz Geni Mutasyon Analizi ... 37
3.3.3. Tam kan sayımı ... 38
3.3.4. Biyokimyasal İncelemeler ... 38
3.4. Organ Tutulumlarının Değerlendirilmesi ... 38
3.4.1. Karaciğer-Dalak Tutulumunun Değerlendirilmesi ... 38
3.4.2. Kemik Tutulumunun Değerlendirilmesi ... 39
3.4.3. Akciğer-Kalp Tutulumunun Değerlendirilmesi ... 40
3.5. Büyümenin Değerlendirilmesi ... 40
3.6. İstatistiksel Yöntem ... 40
4. BULGULAR ... 42
4.1. Zaman incelemesi ... 48
4.1.1 Antropometrik Değişenlerin Zaman İncelemesi ... 48
4.1.2 Labaratuvar Değişenlerinin Zaman İncelemesi ... 50
4.1.3. Görüntüleme Yöntemleri Değişenlerinin Zaman İncelemesi ... 58
4.2 İkili Zaman Karşılaştırmaları ... 60
4.2.1 Antropometrik Değişenlerin İkili Zaman Karşılaştırmaları ... 60
4.2.2. Laboratuvar Değişenlerinin İkili Zaman Karşılaştırmaları ... 62
4.2.3 Görüntüleme Yöntemleri Bulgularının İkili Zaman Karşılaştırmaları .. 64
4.3. Antropometrik Değişkenlerden Boy Z Skorunun Pediatrik Yaş / Erişkin Yaş Gruplarının Karşılaştılması ... 65
4.4. ERT (İmigluseraz) ve ERT (İmigluseraz) / SRT (Miglustat) Gruplarının Karşılaştırılması ... 66
4.4.1. İstatiksel Değişiklikleri Anlamlı Bulunun Antropometrik Değişkenlerin ERT (İmigluseraz) ve ERT (İmigluseraz) / SRT (Miglustat) Gruplarına Göre Tanımlayıcı İstatistik Değerleri .... 66
4.4.2. İstatiksel Değişiklikleri Anlamlı Bulunan Labaratuvar (Hemogram) Değişkenlerin ERT (İmigluseraz) ve ERT (İmigluseraz) / SRT
(Miglustat) Gruplarına Göre Tanımlayıcı İstatistik Değerleri ... 67
4.4.3. İstatiksel değişiklikleri Anlamlı Bulunun Laboratuvar (Biyokimyasal Analiz) Değişkenlerin ERT (İmigluseraz) ve ERT (İmigluseraz) / SRT (Miglustat) Gruplarına Göre Tanımlayıcı İstatistik Değerleri .... 69
4.4.4. İstatiksel Değişiklikleri Anlamlı Bulunun Görüntüleme Yöntemleri Bulgularının ERT (İmigluseraz) ve ERT (İmigluseraz) / SRT (Miglustat) Gruplarına Göre Tanımlayıcı İstatistik Değerleri ... 71
4.5. Splenektomi Olan ve Splenektomi Olmayan Grupların Karşılaştırılması ... 73
4.5.1. İstatiksel Değişiklikleri Anlamlı Bulunun Laboratuvar (Hemogram) Değişkenlerin Splenektomi Olan Ve Splenektomi Olmayan Gruplarına Göre Tanımlayıcı İstatistik Değerleri ... 73
4.5.2. İstatiksel Değişiklikleri Anlamlı Bulunun Labaratuar (Biyokimya) Değişkenlerin Splenektomi Olan Ve Splenektomi Olmayan Gruplarına Göre Tanımlayıcı İstatistik Değerleri ... 75
4.5.3. İstatiksel Değişiklikleri Anlamlı Bulunungörüntüleme Yöntemleri Bulgularınınsplenektomi Olan Ve Splenektomi Olmayan Gruplarına Göre Tanımlayıcı İstatistik Değerleri ... 76
5. TARTIŞMA ... 78
6. SONUÇLAR ... 95
KAYNAKLAR ... 100
EKLER ... 112
TABLOLAR DİZİNİ
Tablo 2.1. Tip 2 Gaucher hastalığının değişik tiplerinde karşılaşılan klinik
bulguların özeti... 11
Tablo 2.2. Enzim replasman tedavisindeki hedefler ... 28
Tablo 2.3. GH Tip I tanısı almış çocuklarda önerilen temel incelemeler ... 31
Tablo 2.4. Nöronopatik Gaucher hastalığı açısından risk grubundaki hastalar ... 32
Tablo 2.5. Nöronopatik Gaucher hastalarında ve risk grubu hastalarda tanı anında yapılması önerilen değerlendirmeler ... 32
Tablo 2.6. GH`da nörolojik tutulumun izlemi ... 33
Tablo 3.1. Olgu formu: Gaucher hastalığı izlemi ... 35
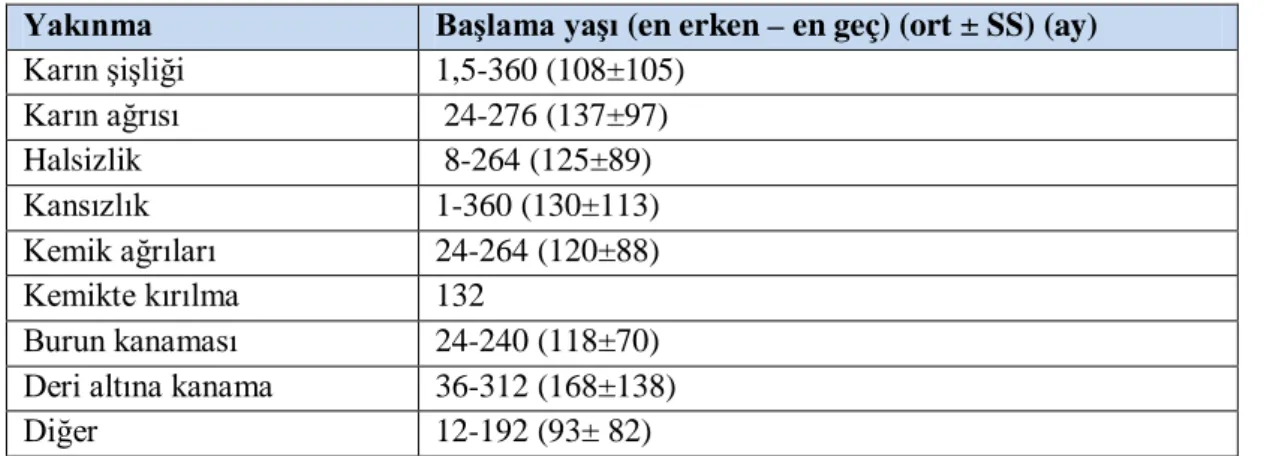
Tablo 4.1. Yakınmaların başlama yaşı ... 44
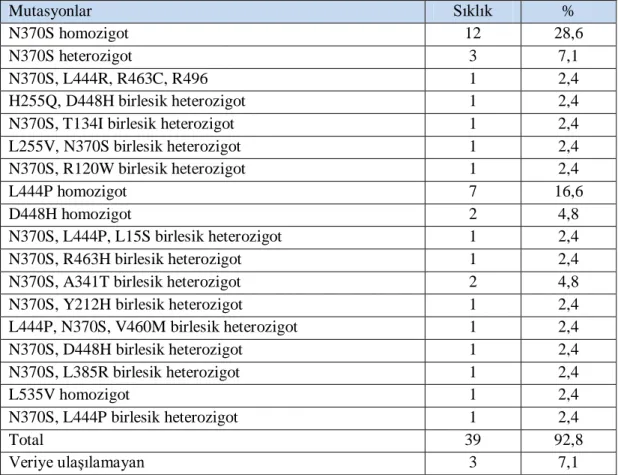
Tablo 4.2. Çalışmaya alınan olgularda hastalığa neden olan mutasyonlar ... 46
Tablo 4.3. Antropemetrik değişkenlerde zaman etkisinin incelenmesi ... 48
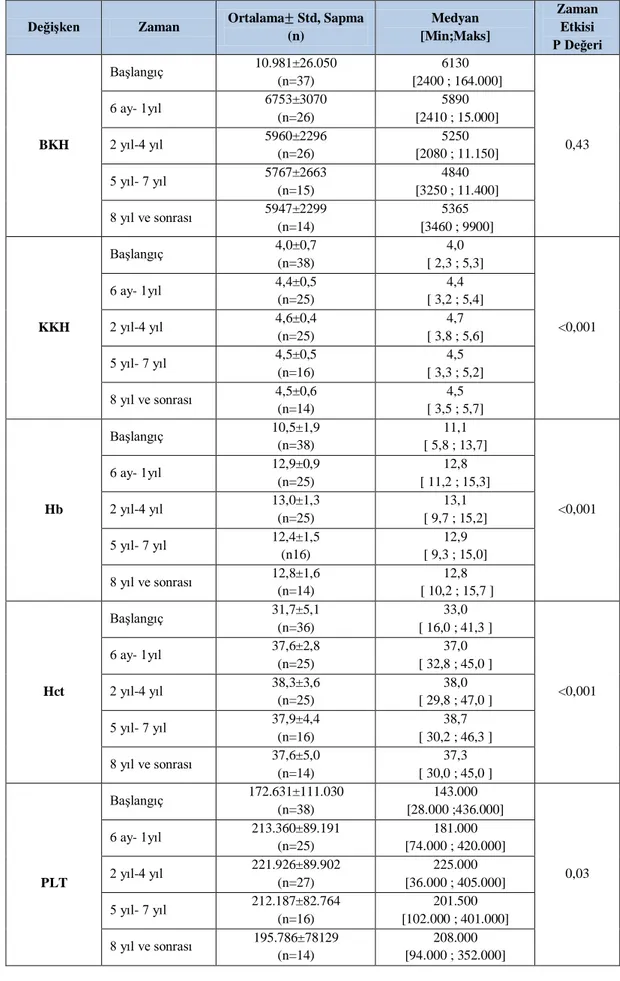
Tablo 4.4. Hemogram değerlerinde zaman etkisinin incelenmesi ... 51
Tablo 4.5. Biyokimyasal değerlerde zaman etkisinin incelenmesi ... 54
Tablo 4.6. Görüntüleme yöntemlerinde zaman etkisinin incelenmesi ... 60
Tablo 4.7. İstatiksel olarak anlamlı bulunan antropometrik değışenlerin ikili zaman karşılaştırmaları ... 62
Tablo 4.8. İstatiksel olarak anlamlı bulunan labaratuar (hemogram) değışenlerin ikili zaman karşılaştırmaları ... 62
Tablo 4.9. İstatiksel olarak anlamlı bulunan labaratuvar (biyokimyasal analizler) değışenlerinikili zaman karşılaştırmaları. ... 63
Tablo 4.10. İstatiksel olarak anlamlı bulunan görüntüleme yöntemler bulgularının ikili zaman karşılaştırmaları. ... 64
Tablo 4.11. Antropometrik değişekenlerin pediatrik ve erişkin yaş gruplarına göre tanımlayıcı istatistik değerleri ... 65
Tablo 4.12. Antropometrik değişekenlerin ERT (imigluseraz) ve ERT (imigluseraz) / SRT (miglustat) gruplarına göre tanımlayıcı istatistik değerleri ... 66
Tablo 4.13. Labaratuar (hemogram) değişekenlerin ERT(imigluseraz) ve ERT (imigluseraz) / SRT (miglustat)gruplarına göre tanımlayıcı istatistik değerleri ... 68
Tablo 4.14. Laboratuvar (hemogram) değişkenlerin ERT (imigluseraz) ve
ERT (imigluseraz) / SRT (miglustat) gruplarına göre tanımlayıcı
istatistik değerleri ... 70
Tablo 4.15. Görüntüleme yöntemleri bulgularının ERT (imigluseraz) ve
ERT (imigluseraz) / SRT (miglustat) gruplarına göre tanımlayıcı
istatistik değerleri ... 72
Tablo 4.16. Labaratuar (hemogram) değişkenlerininsplenektomi olanlar ve
splenektomi olmayanlar gruplarına göre tanımlayıcı istatistik
değerleri ... 74
Tablo 4.17. Laboratuvar (biyokimyavi analizler) değişkenlerinin splenektomi
olan ve splenektomi olmayan gruplarına göre tanımlayıcı istatistik değerleri ... 76
Tablo 4.18. Görüntüleme yöntemleri bulgularının splenektomi olan ve splenektomi
ŞEKİLLER DİZİNİ
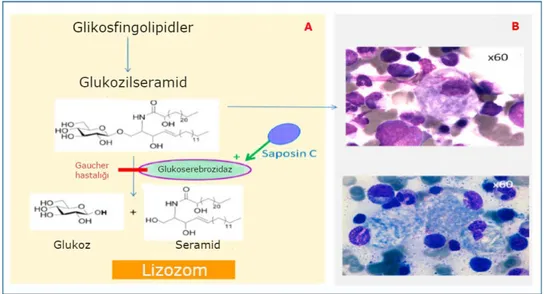
Şekil 2.1. Lizozomda glukosilseramidin (GlcCer) glukoserebrosidaz (GCase)
ile hidroliz edilmesi (A) ve "buruşmuş kağıt mendil" görünümü (B) ... 6
Şekil 2.2. Erlenmeyer Flask deformitesinin röntgen filmi, kortikal inceltme ile birlikte konik bir şişeye benzeyen dia-metafiz birleşiminde eğri eksikliğini gösterir (sağdaki resim) ... 19
Şekil 2.3. Lomber vertebranın T1 ve T2 görüntüleri, vertebra cisimlerinde osteoskleroz değişikliklerini temsil eden kemik iliğinde (oklar) bir azalmayı ortaya koymaktadır ... 19
Şekil 2.4. Bilateral akciğer alanlarında retikülonodüler işaretleri gösteren göğüs radyografisi PA görünümü ... 21
Şekil 2.5. (a-d) HRCT akciğer penceresi aksiyel görüntüler. (b-d) Buzlu cam üst loblar (b) ve bazlar seviyesinde interlobüler ve intralobüler septal kalınlaşma ... 22
Şekil 4.1. Hastaların cinsiyete göre dağılımı ... 43
Şekil 4.2. Hastaların şikayetlerine göre dağılımı. ... 44
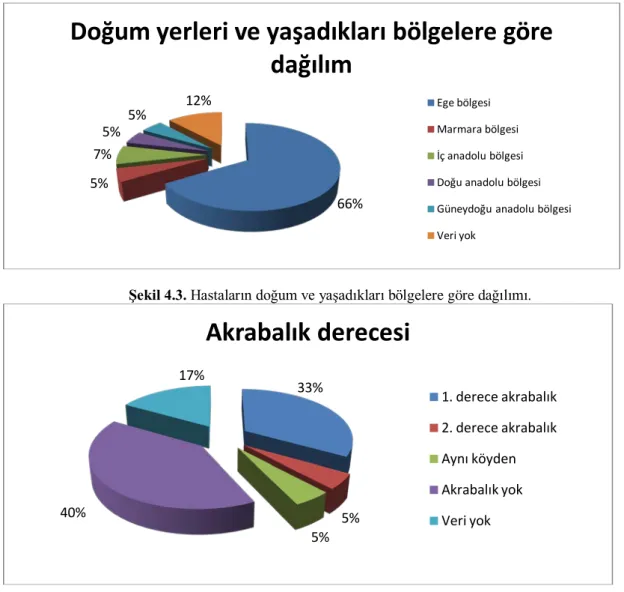
Şekil 4.3. Hastaların doğum ve yaşadıkları bölgelere göre dağılımı. ... 45
Şekil 4.4. Hastaların akrabalık derecelerine göre dağılımı ... 45
Şekil 4.5. Ailede aynı tanı ... 45
Şekil 4.6. Gaucher hastalığı tipleri ... 46
Şekil 4.7. Akciğer tutulumu... 46
Şekil 4.8. Boy ve ağırlık Z skorunun zamana bağlı değişimi ... 49
Şekil 4.9. Femur ve vertebra DEXA`nın zamana bağlı değişimi ... 49
Şekil 4.10 KKH mm3 (A), Hb mm3 (B), Hct mm3 (C) ve PLT mm3 (D)`nin zamana bağlı değişimi ... 52
Şekil 4.11. Ferritin ng/ml (A), albumin gr/l (B), GGT U/l (C), ALP U/l (D), 25 OH Dvit ng/ml (E), PTH pg/ml (F), asit fosfataz U/l (G) ve kitotirosidazın U/l (H) zamana bağlı değişimi ... 58
Şekil 4.12. Karaciğer USG (A), dalak USG (B), karaciğer BT (C) dalak BT (D) femur DEXA(E), vertebra DEXA (F) `nin zamana bağlı değişimi ... 61
Şekil 4.13. Pediatrik ve erişkin yaş gruplarına göre ağırlık z skoru için göreli etkiler ... 65
Şekil 4.14. ERT (imigluseraz) ve ERT (imigluseraz) / SRT (miglustat) gruplarına
göre ağırlık z skoru için göreli etkiler ... 67
Şekil 4.15. ERT (imigluseraz) ve ERT (imigluseraz) / SRT (miglustat) gruplarına
göre KKH (A), Hb (B), Hct (C) ve PLT (D) değişkenleri için göreli etkiler ... 69
Şekil 4.16. ERT (imigluseraz) ve ERT (imigluseraz) / SRT (miglustat) gruplarına
göre albumin (A), GGT (B), alkalen fosfataz (C) ve trigliserit (D) değişkenleri için göreli etkiler... 71
Şekil 4.17. ERT (imigluseraz) ve ERT (imigluseraz) / SRT (miglustat) gruplarına
göre karaciğer (A), femur DEXA (B) vertebra DEXA (C) değişkenleri için göreli etkiler ... 73
Şekil 4.18. Splenektomi olanlar ve splenektomi olmayanlar gruplarına göre
KKH (A), Hb (B), Hct (C) ve PLT (D) değişkenleri için göreli
etkiler ... 75
Şekil 4.19. Splenektomi olan ve splenektomi olmayan gruplara göre albumin
(A), alkalen fosfataz (B) değişkenleri için göreli etkiler ... 76
Şekil 4.20. Splenektomi olan ve splenektomi olmayan gruplarına göre femur
SİMGELER VE KISALTMALAR
ACE Anjiyotensin dönüştürücü enzim
ALT Alanin aminotransferaz
ALP Alkalen fosfataz
ASF Asit fosfataz
AST Aspartat aminotransferaz
BT Bilgisayarlı tomografi
cm Santimetre
DEXA Dual enerji X-ışını abzorpsiyometresi
dl Desilitre
4-MU 4-metilumbeliferon
EEG Elektoensefalografi
FDA “Food and Drug Administration”
GBA Glukoserebrosidaz
gr Gram
GH Gaucher hastalığı
γ-GT Gama glutamil transferaz
HRCT Yüksek çözünürlüklü bilgisayarlı tomografi
Ig İmmünoglobulin
IQ “Intelligence Quotient” (Zeka bölümü)
kg Kilogram
KMD Kemik mineral dansitesi
KMİ Kemik mineral içeriği
L Litre LDH Laktik dehidrogenaz mg Miligram mgr pr Miligram protein ml Mililitre mm Milimetre
MIP Makrofaj inflamatuvar protein
MRG Magnetik rezonans görüntülemesi
nm Nanometre
Nk Normalin katı
SF Serum fizyolojik
SPSS “Statistical Package for Social Sciences”
SSI Semptom ciddiyet skoru
SSS Santral sinir sistemi
TR Triküspid yetmezlik
TRAP Tartarat dirençli asit fosfataz
U Ünite
1. GİRİŞ VE AMAÇ
Gaucher hastalığı (GH), beta glukoserobrozidaz enzim eksikliği ile karakterize, lizozomal lipit depo hastalığıdır. Otozomal resesif kalıtımla geçer. Glukoserebrozidaz enzimini kodlayan genin mutasyonu sonucunda makrofajlarda glukoserebrosid depolanması söz konusudur. Sıklıkla kemik iliği, lenf bezleri, karaciğer ve dalağı tutar. Genel toplumda insidansı 1/40.000-1/60.000 arasındadır 1
. Ülkemizde yapılan bir çalışmada 2,3/1.000.000 sıklık tespit edilmiştir 2. GH`nın 3 alt
grubu vardır:
Tip 1 (kronik nonnöropatik tip), tip 2 (akut nöronopatik veya infantil tip), tip 3 (kronik nöronopatik veya jüvenil tip). Tip 1’de hematolojik bulgular (bisitopeni, pansitopeni), hepatosplenomegali, kemik kırıkları görülür ancak sinir sistemi tutulumu görülmez 3. Tip 2’de hepatosplenomegali, hipersplenizm ve ilerleyici, ağır, sinir sistemi tutulumu olur, hastalar genelde ilk 2 yılda kaybedilirler 4. Tip 3 ise daha ileri yaşlarda görülür ve hepatosplenomegali, kemik, kardiyak ve sinir sistemi tutulumu mevcuttur 5.
Tanıda pansitopeni, karaciğer, dalak veya kemik iliği biopsi bulguları, artmış kitotriozidaz enzim aktivitesi önemli yere sahiptir. Lökositlerde glukoserebrozidaz aktivitesinin %30’dan daha düşük olması ve GBA gen mutasyonlarının tespiti ile tanı konulur 1.
Gaucher hastalığının tedavisinde, eksik enzimi yerine koyma = enzim replasman tedavisi (ERT) temel tedavi yöntemidir. Ancak toksik etkiye sahip madde birikimini engelleyen tedavi = substrat reduksiyon tedavisi (SRT) de seçilmiş hastalarda alternatif tedavi olarak kabul edilmektedir 6. Tartrat rezistan asit fosfataz (TRAP), anjiyotensin converting enzim (ACE), kitotriozidaz hastalığın izleminde ve tedavinin etkinliğinin değerlendirilmesinde önemli yere sahip göstergelerdir 7
.
Enzim replasman tedavisi ve substrat redüksiyon tedavisinin kullanıma girmesiyle hastaların mortalite ve morbiditesi üzerinde önemli yol katedilmiştir. ERT, iki hafta arayla, intravenöz infüzyon şeklinde verilir. Doz genellikle 30U/ kg, ancak 15U / kg ve 120 U/kg arasında değişebilir. SRT ise oral olarak kullanılan preparatlar ile yapılmaktadır 1
.
Bu çalışmada, Ege Üniversitesi Tıp Fakültesi, Çocuk Sağlığı ve Hastalıkları Anabilim Dalı, Metabolizma – Beslenme Bilim Dalı’nda Gaucher hastalığı tanısı ile izlenen hastaların uzun dönem izlem sonuçlarının verilmesi amaçlanmıştır. Buna yönelik
olarak ERT, SRT alan Gaucher hastalarının tedaviye yanıtlarının klinik, labaratuar ve görüntüleme yöntemleriyle retrospektif olarak değerlendirilmesi planlanmıştır.
2. GENEL BİLGİLER
2.1. Tanım ve Tarihçe
Hastalığa 1905’de, bu patolojiye sahip olan hastanın ilk premortem teşhisini koyan Brill tarafından “Gaucher Hastalığı” adı verilmiştir 8
. Philippe Charles Ernest Gaucher 1882 tarihinde karın ağrısı, kanama, anemi, kaşeksi ve hepatospleno- megalisi olan 32 yaşında bir kadın hastanın değerlendirilmesinde, dalaktan köken alan epitelyal hücreler saptamış, bu hücreleri malign epitelyoma olarak değerlendirmiştir. Bu vakanın ilgınç olan yanı, malignite, Gaucher hastalığının klasik klinik bulgularıyla başvurduğu ilk tanıdır; ancak adı geçen Gaucher hücreleri (köpüklü makrofajlar), Gaucher hastalığınına özgür katakteristik hücrelerdir 9
. 1927'de Oberling ve ark. çocukluk formları arasında daha yaygın olan Gaucher hastalığının nörolojik formunu keşfetmiştir. 1960'lı yıllarda Brady ark.10
glikoserobrozid enzim eksikliğini bularak bu hastalığın anlaşılmasına büyük katkılar sağlamışlar ve bu da GH`na neden olan genetik mutasyonun Ginns ve diğerleri tarafından klonlanmasına yol açmıştır. İnsan GBA geninin birinci kromozomda yeraldığı 1983’de Barneveld 11
ve ark. tarafından gösterilmiştir. Takip eden yıllarda genin klonlanması ileç alışmalar artmış, 1996’da Beutler ve Gelbart farklı fenotipik bulgulara sahip Gaucher hastalarında bir çok mutasyon tanımlamıştır 12. Gabrowski ve Pastores gibi bilim adamları, bu hastalığın tedavisinde hastalara enzim replasmanı kullanarak yardımcı olmak konusunda büyük ilerlemeler kaydetmiştir.
Gaucher hastalığı, dünya genelinde görülen en yaygın lizozomal depo hastalığıdır. Otozomal resesif kalıtımla geçer. GH, açıklanamayan hepatospleno-megali ve sitopeni bulguları olan her yaştaki çocuk veya erişkin hastada ayırıcı tanıda yer almalıdır. Glukoserebrozidaz enzimini kodlayan genin mutasyonu sonucunda enzim eksikliğine bağlı makrofajlarda glukoserebrosid depolanması söz konusudur. Sıklıkla kemik iliği, lenf bezleri, karaciğer ve dalağı tutar. Tip 1 nöronopatik olmayan, tip 2 ve 3 ise nöronopatik formlardır. Tip 2 genellikle iki yaşına kadar mortaliteye neden olan daha ağır bir nöronopatik formdur. Tanı, lökositlerde glukoserebrozidaz (beta glukozilseramid) aktivitesinin %30’dan düşük olması ve mutasyonun gösterilmesiyle konulur. Kemik iliğinin histolojik olarak incelenmesi Gaucher hastalığı tanısında yardımcı testlerdir. Mutasyonlar için yapılan
moleküler çalışmalar tanıyı doğrulamak, aile üyelerini taramak ve hastalığın tipi ve prognozu hakkında bilgi edinmek için yararlıdır 13
.
2.2. Epidemiyoloji ve Genetik
Genel toplumda insidansı 1/40.000 - 1/60.000 arasındadır. Hastaların %90 – 95 'inde baskın olarak Tip 1 hastalığına sahip olduğunu göstermektedir. Nörolojik sistemin değişken derecede etkilenmesiyle giden Tip 2 ve 3, çok nadir bir insidansa sahiptir ve popülasyonun 1/100.000'den azında görülür. Tip 1 Gaucher hastalığı çoğunlukla yetişkinlerde görülür ve en yaygın lizozomal depo hastalığıdır. Aşkenazi yahudilerinde GH prevalansı yahudi olmayan papulyasyona gore çok daha yüksektir (118/100.000). Aşkenazi yahudileri arasındaki taşıyıcılık frekansı %6 olduğu halde yahudi olmayanlarda bu oran %0,8'e beraberdir 14. Ülkemizde yapılan bir çalışmada 2,3/1.000.000 sıklık tespit edilmiştir 2.
Bu nadir hastalığın gerçek insidansının belirlenmesi, tedavinin erken başlanması kritik önem taşır. Yenidoğan taraması, erken tanı ve GH'nin nispeten doğru insidansını sağlar. Aldemir O. Ve ark.15 tarafında 2013 yılında Ege
Üniversitesinde yapılan çalışmada, yenidoğan kurukan örneklerinde α-Glikosidaz, β-glikosidaz ve galaktosidaz aktiviteleri florometrik olarak tespit edilmiştir. α-Galaktosidaz aktivitesi kızlarda erkeklere kıyasla daha yüksek olduğu ve ilginç bir şekilde, 38 haftadan önce doğan yenidoğanların α-glikosidaz ve β-glikozidaz aktivitelerinin 39-40 haftada doğanlardan anlamlı derecede düşük olduğu belirlenmiş. Kang.L ve arkadaşları 16 tarafından yapılan diğer bir çalışmada kuru
kanda asit β-glukoserebrosidaz (GBA) aktivitesini saptamak için bir fluorometrik yöntem geliştirilmiştir. Bu çalışma da GBA aktivitesinin florometrik analiz ile yenidoğanlarda yapılan taramasının, verimli ve uygulanabilir bir teknoloji olduğu gösterilmiştir.
GBA geni kromozom 1q21 üzerinde lokalizedir, 7,6 kb büyüklüğündedir ve 11 ekzon ve 10 introndan oluşur 17. Nokta mutasyonları (missense and nonsense), delesyonlar ve eklemeler, junction mutasyonları ve rekombinant alelleri içeren GBA geninde 420'den fazla mutasyon saptanmıştır. Yahudi kökenli Gaucher hastalarında N370S (c.1226A> G) en sık rastlanan mutasyon olarak bildirilmiştir. Bu mutasyon dışında sık raslanan mutasyonlar içinde daha çok öne çıkan mutasyonlar: L444P, 84GG, IVS2+1 mutasyonlardır ve yahudi popülasyonunda mutant allellerin %90’ nı
oluşturmaktadır, diğer beyaz ırklarda bu mutasyonlar mutant allellerin %75’inden azını oluşturmaktadır. En sık görülen mutasyon olan N370S mutasyonunun GH tip1 ile ilişkili olduğu bilinmektedir. Sık görülen mutasyonlardan biri olan homozigot L444P’nin nöronopatik tip GH ileilişkisi gösterilmiştir. Homozigot D448H mutasyonu ise aort ve mitral kapak kalsifikasyonları ile karakterize kardiyovasküler tutulumun ön planda olduğu, okulomotor apraksi, korneada görülen opasitelerin eşlik ettiği bir klinik tipe yol açmaktadır 17, 18
2.3. Patofizyoloji
GBA1 genindeki mutasyonlar, glikoserebrosidaz aktivitesinde belirgin bir düşüşe yol açar. Bu eksiklik sonucu olarak glikoserobrosid makrofajlarda birikir ve bu makrafajların Gaucher hücrelerine dönüşmesiyle sonuçlanır. Işık mikroskopisi altında, Gaucher hücreleri genellikle eksantrik çekirdekler ve yoğunlaştırılmış kromatin ve sitoplazma ile heterojen "buruşmuş kağıt mendil" görünümündedir (şekil 2.1). Bu özellik, elektron mikroskopisi kullanılarak görülebilen karakteristik kıvrılmış, fibriler dizilişdeglikoserobrosid birikimi ile ilgilidir 19
. Gaucher hücreleri ağırlıklı olarak kemik iliği, dalak ve karaciğerde birikir, ancak diğer organlara da birikmektedir ve hastalık semptomlarının başlıca faktörleri olarak kabul edilir. Makrifajlarda glikoserobrosid birikmesi, kemik tutulmasına yönelik ilk adım olarak kabul edilir ve nekrotik komplikasyonların kaynağı olan vasküler kompresyona yol açar 20. Nörolojik tutulumun patofizyolojik mekanizmaları açıklanamamıştır; nöronlardaki glikoserobrosid birikimi sadece rezidüel glikoserebrosidaz aktivitesi büyük ölçüde azaldığında, yani yalnızca bazı GBA mutasyonları tiplerinde önemlidir. Buna uygun olarak Drosophila modelinde yapılan son çalışmalar, glikoserebrosidaz eksikliği olandeney hayvanlarının beyinlerinde otofaji bozukluğu olduğunu gösterilmiştir 21
.
Enzim eksikliği dışında aktive edici protein olan saposin C defekti PSAP geninde mutasyon sonucu oluşmaktadır. Klinik GH tip 3 ile benzerdir 22
Şekil 2.1. Lizozomda glukosilseramidin (GlcCer) glukoserebrosidaz (GCase) ile hidroliz edilmesi (A) ve "buruşmuş kağıt mendil" görünümü (B) 13
. 2.4. Tip 1 Gaucher Hastalığı
GH tip 1 genellikle nörolojik tutulumun yokluğu ile ayırt edilen ve hastalığın en yaygın şekli olan tipidir (Avrupa'da ve Kuzey Amerika'da %90-95). Bu tipin klinik spektrumu değişkendir. Asemptomatik formlardan başlayarak çocukluk çağında ortaya çıkan erken başlangıçlı formlar arasında değişir. Başlangıçtaki semptomlar önemli ölçüde değişkendir ve hastalar her yaşta teşhis edilebilir 23
. Tanının medyan yaşı 10-20 yaş aralığındadır. Bu grubun üçte ikisi (%68) 10 yaşından önce ve neredeyse yarısı (%48) 6 yaşından önce teşhis edilmektedir. GH tip 1 sıklıkla yaşam kalitesini sınırlar ve genellikle ciddi morbiditeye neden olur, ancak nadiren yaşamı tehdit eder niteliktedir3, 24
. Hastaların %50'sinde yorgunluk yakınması mevcuttur ve genellikle okul yaşantısı veya sosyo-mesleki etkinlikler üzerinde bir etkisi vardır24
.
Splenomegali, hastaların %90'ından fazlasında görülmekte ve bazen çok büyük olup dalak birkaç kilograma ulaşabilmektedir. Bu da yaygın karın ağrısı veya distansiyona neden olmaktadır. Splenik infarktüs ve dalak rüptürü çok nadiren görülür25, 26.
Hastaların %60 - 80'inde hepatomegali görülür. Fibrozis ve izleyen siroz gelişimi nadirdir. Akut karın ağrısı ile ortaya çıkan karaciğer ve dalak enfarktüsü gözlenebilir 23.
GH tip 1 hastalarının %40 kadarında karaciğer ve / veya dalakta gaucheroma adı verilen fokal lezyon saptanabilir. Gaucheroma gaucher hücrelerinin çeşitli hücrelerde birikmesinden oluşan fokal lezyondur. Genellikle karaciğer USG`sinde tesadüfen saptanmaktadır. Gaucheromalar çeşitli görüntüleme özelliklerine sahiptir ve bu nedenle bir gaucheromayı başka bir lezyontan ayırmak zordur ve hepatoselüler karsinom veya bir lenfoma ile karışabilir27
.
Safra taşlarının GH tip 1'deki prevalansı %32'dir, yani genel popülasyondan beş kat daha fazladır28
.
Değişken derecelerde trombositopeni yaygındır (vakaların%90'ı). Anemi daha az yaygındır (vakaların%20-50 'sı) ve orta derecedir, hemoglobin seviyeleri nadiren 9 g/dL'den düşük bulunmuştur. Lökopeni nadirdir. Trombositopeni olmayan GH vakaları da mevcuttur. Bu sitopeni, splenik sekestrasyon ve kemik iliği infiltrasyonu ile ilişkilendirilir, ancak olgunlaşmamış hematopoietik hücreler üzerindeki enzim eksikliğinin doğrudan etkisi tarif edilmiştir. Splenektomi öyküsü olan hastalarda kan sayımı normal olabilir. İmmün trombositopeni de tanımlanmıştır29,30
.
GH'da, protrombin süresi (PT) uzamış ve aktive kısmi tromboplastin zamanı (APPT) dahil olmak üzere çeşitli hemostatik anormallikler tanımlanmıştır. Bunlar muhtemelen faktör X, faktör V, trombin (veya daha global bir eksiklik), faktör XI veya K vitamini eksiklikleri,karaciğer yetmezliğine sekonder veya potansiyel olarak genetik veya hatta edinilmiş von Willebrand hastalığı ile ilişkili olabilir 30
.
Kemik tutulumu, ağırlıklı olarak pelvis ve alt ekstremitede (daha nadiren üst ekstremitede) akut ağrılı kemik krizleri ve ya kronik ağrı görülebilir. Ağrının şiddeti değişkendir ve prognoz üzerinde etkisi mevcuttur31
. Kemik belirtilerinin patofizyolojisi çok iyi bilinmemektedir. Ağrılı kemik krizi muhtemelen iskemik vaso-okluzif olaylarla ilişkilidir. Genellikle uzun kemiklerde (metafizler veya diyafizler) ve düz kemiklerde kemik infarktları ve epifizlerde avasküler nekroz (AVN) olarak adlandırılan lezyonlara neden olurlar. İskemik olayları (kemik enfarktüsü ve AVN) açıklayan damar teorisine ilaveten, özellikle femur başıda, spontan veya trabeküler mikrokırıkları (mekanik veya spontan AVN) açıklamak için mekanik bir teori öne sürülmüştür. Patofizyolojik mekanizma, kemik iliği veya immün hücrelerdeki değişiklikler, inflamasyon, makrofaj kaynaklı faktörler, sitokinler ve hormonlar gibi diğer potansiyel faktörleri de içerir 32
.
Akut ağrılı kemik krizi çocuklarda daha sıktır (GH tip 1 olan çocukların %30'u). Genellikle 7 - 10 günden fazla sürmektedir ve lokal inflamasyon, hafif ateş
(38°C), polinükleer lökositoz ve ılımlı bir inflamatuar sendrom ile ilişkilendirilmektedir. Bu semptomlar, osteomiyelit (psödoo-osteomiyelit) ile benzerdir, bu nedenle bazen tanı gecikir 31, 33. AVN vakaların %15'inde, çoğunlukla femur veya humerus başında, nadiren femur kondillerde veya tibia platolarında ve bazen ayak bileğinde (talus, kalkaneus), ellerde ve omurgada (vertebra plana) görülür. Uzun dönemde AVN, osteoartrit nedeniyle komplike olabilir, sıklıkla eklem replasman cerrahisini gerektirir34.
Osteopeniveya osteoporoz, sağlıklı bireylerde yaş ve menopozla birlikte artmaktadır. Gaucher hastalarında kemik kitlesi kaybı daha erken meydana gelir ve daha şiddetli olup, patolojik kırıklara (uzun kemikler, vertebra vb.) neden olabilir. Kemik kitlesi azalması, diğer kemik ve visseral komplikasyonlar ile korelasyonlu gibi gözükmektedir 35. Kortikal kemiği aşındırabilen ve patolojik fraktürleri teşvik edebilen fokal litik lezyonlar bazen farklı yerlerde (uzun kemikler, çene vb.) görülür. Trabeküler yapı kaybı ile mandibuladaki kist benzeri lezyonlar diş anormalliklerine neden olabilir 36. Osteosarkomlar veya osteoblastomlar dahil olmak üzere sekonder kemik tümörleri çok nadir olarak bildirilmiştir 37, 38
.
Çocukluk döneminde ortaya çıkan Erlenmeyer flask deformitesi olarak adlandırılan, femur alt kısmının metafiz / diyafizal bölgesinin genişlemesi ile kemik remodeling bozukluklarını gözlemlemek için standart radyografiler kullanılabilir. AVN ve kemik enfarktüsünün sekeli, kortikal kemiğin incelmesi, fokal lizis, kırıklar ve osteoartirit gözlenebilir. Kemik iliği infiltrasyonu (olguların %80'inde), kemik enfarktüsü ve AVN`u çok erken bir aşamada belirlemek için MR kullanılabilir 39. Akciğer tutulumu, Gaucher hücreleri tarafından akciğerlerin infiltrasyonu ile ilişkili olabilir ve pulmoner fibrozise neden olabilen bir interstisyel hastalıktır. Spinal deformasyona sekonder restriktif akciğer hastalığı veya pulmoner arteriyel hipertansiyon da görülmektedir 40, 41. İkincisi, splenektomili hastalarda, özellikle kadınlarda daha sıktır veya karaciğer sirozu ile komplike olan hepatopulmoner sendromdan kaynaklanabilir. Akciğer tutulumu nadirdir ve homozigot L444P mutasyonlu hastalarda daha sık gözükmektedir 42
.
Nadiren, proteinüri ve hematüri olarak ortaya çıkan böbrek tutulumu, glomerüllerin Gaucher hücreleri tarafından infiltrasyonunu yansıtmaktadır 43. Deri tutulumu sarı rengli hiperpigmentasyon şeklinde, genellikle tibiaların ve yanakların ön kısımlarında görülür 44
vitrioretinal tutulum (glukoseramid yataklara karşılık gelen), miyokardiyal veya kapak tutulumu, insülin direnci ve amiloidoz bulguları çok nadiren gözlenir45, 46, 47, 48.
GH tip 1'in konvansiyonel tanımının aksine, bu fenotipe bağlı bazı nörolojik bulgular son yıllarda açıklanmıştır. GH tip 1 hastalarının parkinson hastalığına yakalanma riski artar (4-20 kat daha fazla) ve bu yakalanma klasik parkinson hastalığından daha erken yaşlarda görülmektedir 49
Minimal semptomatik periferik nöropatilerin prevalansı %14'tür ve bu nedenle genel popülasyondan daha yüksektir50.
2.5. Tip 3 Gaucher Hastalığı
GH tip 3, kronik progresif nöronopatik tip olarak adlandırılır ve bebeklik, çocukluk, ergenlik veya yetişkinlik döneminde bulgu verebilir (tüm olguların %5, ancak bazı kohort çalışmalarında %33'e kadar olduğu belirtilmektedir.) 51
. GH tip 1'de tanımlanan visseral bulgulara ek olarak okulomotor, nörolojik tutulum da mevcuttur. Genellikle 20’li yaşlarda görülmekte olan hastalığın patogenezi özellikle nörolojik bulgular açısından heterojendir. Bazı vakalarda tek nörolojik semptom olarak horizontal oftalmopleji gibi orta dereceli sistemik tutulum gösterirken, bazılarında ilerleyici miyoklonus, epilepsi (hastaların %16'sı), serebellar ataksi veya spastisite (hastaların %20 - 50'sinde), bunama gibi değişik nörolojik bulgular göstermektedir. Bu nörolojik bulgular, visseral bulguların ortaya çıkmasından birkaç yıl sonra başlangıçta GH tip 1 olarak tanımlanan ve tedavi edilen hastalarda bile ortaya çıkabilir 52, 53
.
Bazı Gaucher hastalarında tedaviye rağmen bilinmeyen bir mekanizma ile kifoz, kardiyak tutulum (kapak kalsifikasyonu ile), korneal tutulum ve hidrosefali de görülmektedir54. Kardiyak tutulum (kapak kalsifikasyonu ile), korneal tutulum ve hidrosefali, esas olarak c.1342G> C (D448H) genotipi olan GH tip 3 hastalarında rastlanmıştır55
.
Gaucher hastalarının yer aldığı, uluslararası kayıt sisteminden önce, GH tip 3 hastalarının en büyük çalışmasını Dreborg ve ark.56
yapmıştır. Çalışmada Norrbottnian kökenli, yaş aralıkları 8 ay ile 14.5 yıl arasında olan 22 hasta bildirmiştir. Bu çalışmada ilk bulguların ortaya çıkma yaşı ortalama 12 ay, klinik olarak başlangıçta normal zekaya sahip olan hastalarda hepatosplenomegali, boy kısalığı, kaba motor gelişmede %30 ve ince motor gelişmede %10 gecikme
saptanmıştır. Tanıda, %25 hastada strabismus ve ya anormal göz hareketleri mevcuttur.
Daha sonra bir diğer çalışma Gaucher hastalarının yer aldığı, uluslararası kayıt sistemi tarafından yapılmıştır. Çalışma sonunda bu hastalığın önceleri düşünüldüğün-den daha sık olduğu ve tüm etnik gruplarda görüldüğü anlaşılmıştır. Çalışmada 400'den fazla etnik ve demografik açıdan farklı GH tip 3 hastası incelenmeye alınmıştır. Hastaların çoğunluğu Mısırdandı (%31) ve L444P/L444P genotipi sıklık oluşturuyordu. Diğer genotiplerse %6-8 oranla L444P/D448H, D448H/D448H’di. Göz hareketi anormallikleri genellikle ilk semptom olarak kendini göstermekteydi ve hastaların %60'ında mevcuttu. Motor nöron anormallikleri, ataksi, spastisite, miyokloni, nöbetler hastalıkların seyrinde ortaya çıkmıştı. Genel olarak, beyin sapı ve ince motor disfonksiyon en sık görülen nörolojik bulgulardi. Hastalarda tanı anında gelişimsel gecikme % 43 idi52
.
GH tip 3, klinik olarak 3 alt tipe ayrılmıştır. Tip 3a belirgin nörolojik bulguların yanında hafif viseral tutulumun olması ile karakterizedir. Daha çok erken çocukluk döneminde myoklonus, demans ve ataksi gibi nörolojik bulgular ile kendini gösterir. Tip 3a’nın en önemli özelliği erken dönemde, horizontal sakkadik göz hareketlerinin yavaşlaması ile karakterize yatay supranükleer bakış paralizisinin ortaya çıkmasıdır. Bu hastalar daha ileri dönemlerde, zayıf göz hareketlerini, sinkinetik göz kırpma ve kafayı çevirme ile telafi edebilmektedir. Tip 3a’nın seyrinde tedaviye dirençli jeneralize tonik - klonik nöbetler, demans, ilerleyici spastisite, ataksi gelişir ve sonuçta hasta entelektüel işlevlerini yitirir. Nörolojik dejenerasyon bu hastaların ikinci veya üçüncü on yılda ölümüyle sonuçlanır. Bu hastalarda karaciğer ve dalak genellikle büyümüştür. Kemik tutulumu çoğu hastada mevcuttur. GH tip 3b bebeklikte ve erken çocukluk döneminde ortaya çıkar, belirgin hepatosplenomegali, kanama eğiliminde artma, kemik tutulumu, akciğer tutulumu ve büyüme geriliği gibi agresif sistemik özellikler ile kendini gösterir. Bu hastaların nörolojik tutulumu genellikle erken başlayan supranükleer bakış paralizi ile sınırlıdır ancak nöbetler görülebilir .Tip 3c olarak adlandırılan klinik tipte, supranükleer bakış paralizi, hafif hepatosplenomegali ve kemik tutulumu, korneal opasiteler, mitral ve aort kapak kalsifikasyonu mevcuttur. Tip 3c homozigot D448H mutasyonu ile birliktedir 57.
2.6. Tip 2 Gaucher Hastalığı
1927'de tanımlanmlanan GH tip 2, gaucher hastalığının nadir ve en şiddetli formudur. GH tip 2 genellikle prenatal dönemde veya bebeklik döneminde semptomlar gösterir ve genellikle 3 yaşından önce ölümle sonuçlanır 4. GH tip 2, GH'nın en az görülen formudur. Genel olarak, 100.000 ila 500.000 canlı doğumda bir tahmini bir sıklığa sahiptir 58
. Opistotonus, bulbarbulgular (özellikle şiddetli yutkunma bozuklukları) ve okülomotor sinir felcini (veya bilateral sabit şaşılık) içeren üçlü, GH tip 2 için karakteristiktir. Bu bulguların trismus, piramidal ve muhtamelen ekstrapiramidal hipertoni ile ilişkili olduğu düşünülmektedir 4
. GH tip 2 nin 3 alt tipi mevcuttur 59. (Tablo 2.1)
Tablo 2.1. Tip 2 Gaucher hastalığının değişik tiplerinde karşılaşılan klinik bulguların özeti. 4
Perinatal letal Non-perinatal letal Intermediate
Yaşam süresi Prenatal dönemden 3. aya
kadar 2 yaşa kadar 2-6 yaş
Cilt Hidrops fetalis, iktiozis
Elektron mikroskopisinde stratum korneumun ultrastrüktürel değişiklikleri
Akciğer Pulmoner hipoplazi Tekrarlayan aspirasyon pnömonileri Tekrarlayan aspirasyon pnömonileri, restriktif akciğer hastalığı Visceral organlar HSM, sarılık, GİS kanamaları HSM, GİS kanamaları HSM
Hematolojik Purpura, trombositopeni Anemi, trombositopeni Anemi, trombositopeni
Nörolojik
Artrogrypozis, mikrosefali, emmeme, laringeal stridor, boyun hiper-ekstansiyonu, opistotonus,
apne
Trismus, laringeal stridor, boyun hiperekstansiyonu, özofagus dismotilitesi, hipotoni, atetoz, epilepsi, miyoklonus, apne
Miyoklonik epilepsi, ilerleyici beyin sapı tutulumu, nöbetler
Oftalmolojik
Strabismus, okculomotor apraksi, gözyaşı azlığı /yokluğu Strabismus, Okulomotor apraksi, gözyaşı azlığı /yokluğu Dismorfoloji Düşük kulak, burun küçük ve kökü basık, antevert burun deliği
2.7. Tanı
GH ile uyumlu klinik özelliklere sahip bir hastada, genellikle periferik lökositlerdeki glukoserebrosidaz aktivitesinin azalmış olduğunun gösterilmesi tanıda ilk basamaktır. Mutasyon analizi, tanının doğrulanması ve klinik bulguları önceden tahmin etmede önemlidir. Ayrıca tanısı konmamış aile üyelerini ve heterozigot taşıyıcıları tanımlamada da yol göstericidir 18
.
Enzim aktivitesi, her bir beyaz kan hücre tipinde değişir ve monositlerden lenfositlere, granülositlere doğru azalır. Teşhis aynı zamanda deri biyopsisi ile elde edilen kültür fibroblastlarda veya glukoserebrosidaz aktivitesinin ölçülmesi ile de yapılabilir. Periferik lökositlerde bakılan testde suni bir substrat olan 4 – metilumbilliferil – beta - glukosid kullanır. Bu testle, tip 1 hastalar genel olarak zayıf enzimatik aktivite gösterirler (kontrol enzim aktivitesinin %10 -15'i). Tip 2 ve 3 hastaların enzimatik aktivitelerinin genellikle daha düşük olmalarına karşın birbirlerinden enzim analizi ile ayırd edilemez. Heterozigot taşıyıcılardaki ve normal bireylerdeki enzim aktivitesi bir-birine benzerdir. Dolayısıyla, taşıyıcıları taşıyıcı olmayanlardan ayırmak için enzim analizi tek başına kullanılamaz60
.
Hastalara anemi, trombositopeni ve/veya splenomegali bulguları nedeni ile yapılan kemik iliği aspirasyonunda Gaucher hücrelerinin görülmesi ile tanı konulmasına sıklıkla rastlanır. Ancak lösemi, lenfoma gibi hematolojik malignite-lerde yüksek orandakı yapım ve yıkımın sonucu olarak mikroskopik olarak Gaucher hücreleri ile birebir aynı olan ‘psödo-Gaucher hücreleri’ görülebilmekte, bu da tanı karışıklığına sebep olabilmektedir. Sonuç olarak kemik iliği aspirasyonu, invaziv olması ve spesifik olmayabileceği için ve özellikle enzim aktivitesi tetkikinin kolaylıkla yapılabildiği yerlerde tanı testi olarak önerilmemektedir. Kemik iliği aspirasyonu dışında bazen hastalar dalak, karaciğer, akciğer gibi dokularda Gaucher hücrelerinin görülmesi ile de tanı alabilirler 61
.
Kesin tanı GBA geninde mutasyonun gösterilmesiyle konmaktadır 1
. GBA geni (7.5 kb), 11 ekzon ve 10 introndan oluşur ve birkaç gen içeren kromozom 1q21 loküsünde bulunur. GBA geninde ikiyüzden çok mutasyon gösterilmiştir 62
, Türk toplumunda L444P ve N370S mutasyonlarının sırasıyla mutasyonların %42 - %30'unu oluşturmaktadır. N370S mutasyonunun hafif fenotipten (GH tip 1) sorumlu olduğu ve L444P mutasyonunun ağır fenotipten (GH tip 2 ve 3) sorumlu olduğu bildirilmiştir 63
Karaca ve ark.18 tarafından Ege Üniversitesinde yapılan bir çalışmada çalışmaya alınan 32 hastanın %50`de N370S mutasyonu saptanmış ve bu mutasyonun hem homozigot hem de heterozigot formlarının GH tip 1 ile ilişkili olduğu gösterilmiştir. Hastaların %35,5 saptanan L444P mutasyonu tüm tip 2 ve tip 3 gaucher hastalarında homozigot olarak saptanmıştır. Bu çalışmada L385R yeni mutasyonu keşfedilmiş, bu mutasyon H255Q mutasyonu ile heterozigot olarak bulunmaktadır.
Tanı anındaki labaratuar analizlerinde hemoglobin konsantrasyonu, trombosit sayısı ve biyokimyasal belirteçler (kitotirosidaz ve kemokin [C-C motifi] ligand 18 / pulmoner ve aktive düzenlenmiş kemokin [CCL18 / PARC]) değerlendirilir. Hastalık progresyonunun izlenmesinde de CCL18 / PARC ve kitotriozidaz gibi biyokimyasal belirteç kullanılabilir. Mutlak konsantrasyonlar yararlı değildir, ancak seri artışlar klinik tekrarlamanın erken bir göstergesi olabilir ve hastalığın durumunun ve tedaviye uyumun araştırılmasına yardımcı olur. Kitotirosidaz, Gaucher hücresi tarafından eksprese edilen "alternatif" tip makrofaj aktivasyonunun bir işaretidir. Gaucher hastalarında aktivitesi artar ve klinik yanıtın diğer göstergeleriyle birlikte enzim replasman tedavisi sonrası düzeylerinde azalma olur. Kitotirosidaz ölçümü, başlangıçta ve periyodik olarak tedavi sırasında önerilmektedir. Kemokin CCL18 / PARC alternatif olarak aktive olan makrofajlarda yükselir, bu proteinin konsantrasyonları, visseral ve kemik hastalığının çeşitli yönleriyle ilişkilidir 64
. Glukozil sfingozin de tedavi izleminde kullanlmaktadır. Ayrıca Gaucher hücrelerinin çevresindeki fagositlerden kaynaklanan makrofaj inflamatuvar protein (MIP) 1β ve 1α’nın da Gaucher hastalarının serumlarında yüksek düzeylerde bulunduğu gösterilmiştir. Özellikle MIP 1β’nın kemik tutulumu olan hastalarda daha yüksek olduğu görüldüğünden, kemik tutulum belirteçi olarak kullanılmasına ilişkin çalışmalar devam etmektedir 65, 66
.
2.8. Hastalığın Etkilediği Doku ve Organ Sistemleri
2.8.1. Hepatosplenomegali
Dalak: Splenomegali, Gaucher hastalarının büyük çoğunluğunda
bulun-duğundan, hematolojik, kemik anormallikleri ve diğer patolojik parametreler olsun ya da olmasın çok önemli bir bulgudur. Erken başlangıçlı hastalıkta karın
distansiyonuna yol açarak pelvise kadar uzanabilmekle aynı zamanda kosta kenarında da palpe edilebilir. Splenomegalisi olan hastalarda distansiyon ve karın ağrısı kapsül gerilmesine bağlı olarak oluşmaktadır. Normal dalak volümü toplam vücut ağırlığının %0.2’si olarak tanımlanmaktadır 25. Uluslararası Gaucher Grubu Kayıt sistemindeki verilere göre, hastaların %85'sinde splenomegali normalin beş katından fazladır. Gaucher hastalığı splenomegali olmadan ortaya çıkabilir ve splenomegali yokluğu Gaucher hastalığını dışlatmaz. Genellikle splenomegali hafif olan ve mevcut olmayan olgularda homozigot N370S mutasyonu görülmektedir. Gaucher hastalığında splenektomi karaciğer, iskelet ve akciğer bulgularının agresif seyriyle pozitif ilişkilidir. Bu nedenle, splenomegali nedeni tespit edilemediğinde, splenektomi düşünülen herhangi bir hastada Gaucher hastalığı düşünülmeli ve dışlanmalıdır 24
.
Organvolümü ölçümü için bilgisayarlı tomografi (BT) ve ya tercihen volumetrik magnetik rezonans görüntülemesi (MRG) önerilmektedir, ancak ultrasonografi (USG) ile de deneyimli bir uygulayıcı varlığında tahmin edilebileceği belirtilmektedir 67.
Gaucher hastalığında splenomegali değerlendirildiğinde, dalak parankiminin incelenmesi de yapılmalıdır. Gaucher hastalığında fokal splenik lezyon neredeyse %20 oranında görülmekte ve insidansı dalak büyüklüğüne göre artmaktadır. Bu lezyonlar, Gaucher hücrelerinin fokal koleksiyonu veya enfarktüs alanları olabilir ve dalak neoplazmı olarak yanlış tanıya sebebiyyet verebilir.
Karaciğer: Gaucher hastalarında karaciğer hastalığını değerlendirmek ve
yönetmek zor olabilir. Hepatomegali hastalarda karında distansiyon, şişlik ve erken doyma gibi yakınmalara neden olmaktadır. Normal karaciğer volümü toplam vücut ağırlığının %2.5’i olarak tanımlanmaktadır. Hepatomegali normal karaciğer volümünün 1,25 katı olarak tanımlanır 68
. GH`da substrat birikimi sinüzoidal makrofajlarda meydana gelir ve hepatositler genellikle korunur, ancak belirli oranda depolanma görülebilir. Hepatoselüler işlev korunmuş olsa da karaciğer fonksiyon testlerinde hafif yükselmeler mevcut olabilir .
GH`da karaciğer tutulumu yaygın olmakla birlikte, karaciğer sirozu, portal hipertansiyon dahil olmakla ağır komplikasyonlara neden olan geniş bir klinik yelpazeye sahiptir. Gaucher hastalarında, kolelitiyazis sıklığının artmış olduğu Literatürde GH’nda hepatoselüler karsinom riskinin artmış olduğunu
bildiren yayınlar da mevcuttur. Bununla birlikte GH`nın kendisi de karaciğer lezyonlarını taklit edebilir, karaciğer hastalıklarına özgün laboratuvar testlerini etkiler ve sirotik olmayan portal hipertansiyon ile ilişkili olabilir ve bu da tanı yaklaşımını daha da karmaşık hale getirir. Bu nedenle GH`da karaciğer tutulumunu bilmek, hastalara gereksiz invaziv testler yapılmasını önler ve önemli karaciğer hastalığı şüphesi bulunan hastaları değerlendirirken karar verme konusunda bizlere yardımcı olabilir 69
.
GH spesifik tedavisi mevcut olmadan önce splenektomi yapılan hastalarda karaciğer çok büyük ölçülere ulaşmaktaydı (normalin 10 kat büyüklüğü). Bu splenektomili hastalarda karaciğer sirozu bildirilmiştir. GH da karaciğer fonksiyon testleri genellikle normal aralıkta olmakla birlikte, splenektomili hastalarda ciddi anormallikler görülme olasılığı daha yüksektir 70
.
2.8.2. Hematolojik Değişiklikler
Anemi, trombositopeni ve daha az ölçüde lökopeni, splenik sekestrasyonun da katkısıyla, kemik iliği infiltrasyonunun bir sonucudur ve eşzamanlı veya bağımsız olarak gözlenebilir 71. Uluslararası Gaucher kayıt sisteminde yer alanhastaların %36'sının ilk bulgusu anemidir. Bununla birlikte, splenomegali ile sitopeninin derecesi arasında doğrudan bir korelasyon bulunmamakla birlikte, diğer mekanizmaların da bu bulgulara neden olabileceğini düşündürmektedir 72
. Bunlar, Gaucher hücrelerinin kemik iliğine sızdırılması, demir eksikliği, demir metabolizması ve transportundaki değişiklikler, vitamin B12 eksikliği ve otoimmün hemolitik anemi olabilir 73. Serum ferritin seviyeleri genellikle yükselirken serum demir, transferrin saturasyon katsayısı ve transferrin seviyeleri gibi diğer parametreler normaldir.
Uluslararası Gaucher Kayıt sistemindeki hastaların %15'inde şiddetli trombositopeni (trombosit sayısı 60.000/ml'den az), %45'inde orta düzeyde trombo-sitopeni (trombosit sayısı >60.000'den <120.000/ml) ve %40`ında hafif derecede trombositopeni (Trombosit>120.000 - <150.000 /ml) mevcuttur. Anemi gibi, trombositopeni de hipersplenizm ve / veya kemik iliği infiltrasyonu ile ilişkilidir, splenomegali derecesi ile doğrudan bağlantılı değildir 30
. GH olan hastalar öncelikli olarak ağır trombositopeniye bağlı kanama riski altındadır. Bununla birlikte, kanama eğiliminin derecesinin, trombosit fonksiyonunun ve koagülasyon faktörlerinin eş
zamanlı anormalliklerini düşündüren trombosit sayısı ile orantılı olmadığı kaydedilmiştir. 30 tip 1 Gaucher hastasında pıhtılaşma ve fibrinoliz üzerine yapılan bir çalışmada aktif tromboplastin zamanı (aPTT) ve protrombin zamanı (PT) sırasıyla %42 ve %38 oranında uzamıştır. Trombin-antitrombin kompleksi ve özellikle splenektomili hastalarda fibrinoliz (PAP kompleksi, D-dimer) anlamlı derecede yükselmiştir 74
.
Mukozal kanama ile ilişkili olarak, Tip 1 Gaucher hastalarında önemli ölçüde (Düşük trombosit adezyonu da dahil olmak üzere) trombosit işlev bozukluklarına sahip olduğu bulunmuştur. Yapılan çalışmalar, azalmış adezyonun intrensek bir trombosit defekti olduğunu ve hastalığa özgü ERT'nin kullanılmasından etkilenmediğini göstermiştir 75
.
Gebelik, GH`da kliniği kötüleştirme potansiyeline sahiptir ve doğum sırasında kanama riski Gaucher hastalığı olan kadınlarda artabilir. Bir çalışmada, trombosit agregasyon bozukluğu olan kadınların %78.6'sı en az bir defa doğum sırasında postpartum hemoraji geliştiği belirtilmiştir. Gebelik öncesi ve sırasında enzim replasman tedavisinin spontan düşük ve GH`na bağlı komplikasyonlar, özellikle doğum ve doğum sırasındaki kanama riskini azaltmaktadır 76
.
2.8.3. KemikTutulumu
Kemik tutulumu, ilerlemiş hastalığı olan tedavi edilmemiş hastalar arasında önemli bir morbidite kaynağıdır. Bildirilen komplikasyonlar arasında osteoporoz, vertebra basısı kırığı ve femur başının osteonekrozu mevcuttur 20
. Kemik tutulumu, ağırlıklı olarak pelvis ve alt ekstremitelerde (nadiren üst ekstremitelerde) ağrılı kemik krizleri şeklinde ortaya çıkan akut ağrıya ve / veya kronik ağrıya neden olur. GH da kemik ağrısı yaşam kalitesini önemli derecede etkilemekte. Genel olarak lokalize edilemeyen, sistemik ve lokal bulgular olmaksızın gözlenen ve tedaviye hızlı yanıt veren ağrı gözlenmektedir. Ağrının şiddeti değişken olmakla birlikte fonksiyonel prognoza etkisi mevcuttur 32.
İkinci sıklıkta ise kemik krizleri görülmektedir. Şiddetli ağrıyaneden olan kemik krizi ataklarının standart analjezikler ile kontrol altına alınması mümkün değildir. Yaşanan ağrı ve günlük yaşam aktivitelerinin bozulması yaşam kalitesi üzerine yıkıcı etkiler bırakır. Akut ağrılı kemik krizi çocuklarda daha sıktır (GH tip 1çocukların %30'u). Genellikle 7 - 10 günden fazla devam etmektedir ve lokal
inflamasyon, ateş (38 ° C), polinükleer lökositoz ve ılımlı inflamatuar sendrom ile ilişkilendirilmek-tedir. Bu semptomlar osteomiyelit (psödoo-osteomyelit) ile benzerdir, bu nedenle bazen tanı gecikmektedir 32. AVN vakaların %15'inde, çoğunlukla femur veya humerus başlarında, nadiren femur kondillerinde veya tibia platolarında ve ender olarak ayaklarda (talus, kalkaneus), ellerde ve omurgada (vertebra plana) görülür. Uzun vadede, AVN, osteoartrit nedeniyle komplike olabilir, sıklıkla eklem replasman cerrahisine neden olabilir. Kemik krizleri, ileride olabilecek kemik enfarktüsü ve AVN'yi öngörür. Kemik enfarktüsü, AVN ve patolojik kırıklar GH'nin şiddetli kemik komplikasyonları olarak kabul edilir ve kemik olayları olarak tanımlanır 34
.
Normal kemik kütlesi kayıpları (osteopeni) veya daha şiddetli kayıplar (osteoporoz), normal kişilerde yaş ve menopozla birlikte artmaktadır. Kemik kortekste incelme ve kemik yoğunluğunda azalma, hemen hemen tüm GH`da mevcutur. Trabeküler ve kortikal kemik lokalize ya da yaygın şekilde etkilenmektedir. Osteopeni hem erişkin hem de pediatrik hastada artmış kemik fraktürü riski ile ilişkilidir. Kemik korteksini zedeleyen ve patolojik fraktürlere neden olan fokal litik lezyonlar bazen farklı yerlerde (uzun kemikler, çene vb.) görülür. Kırıklar, sıklıkla femur boynu, torakolomber vertebralarda ve tibiada görülmektedir. Osteonekroz GH’na bağlı kemik hastalığının en ciddi ve sekel bırakıcı komplikasyonudur ve özellikle ikinci on yılda daha sık görülmektedir. Klinik bulguları artraljiden, ciddi eklem destrüksiyonuna kadar değişkenlik göstermektedir. Femur başı en sık etkilenen bölgedir ve genellikle hastalık her iki femur başında da görülmektedir. Daha az sıklıkta femurun distal epifizi, proksimal humerus epifizi ve vertebralar etkilenebilmektedir. Eklem destrüksiyonu sonrası kollaps, genellikle protez takılmasını gerektire bilmektedir. Osteonekroz özellikle osteopeni ile birlikte, bir veya birden fazla vertebranın kırılmasına yol açabilir ve buna bağlı nörolojik bulgular ortaya çıkabilmektedir. Splenektomi, osteonekroz için bir risk faktörüdür. Psödotümor oluşumu ise, GH’nda nadir görülmekte, ancak özgün radyolojik görünümü ve çoğu zaman Gaucher hücrelerinden oluşan yumuşak doku kitlesi ile birlikte olması nedeni ile tanınması gereken bulgulardandır. Splenektomi geçiren hastalarda tüm kemik bulgularında kötüleşme olabilmektedir 32. Kemik iliği hastalık
yükünü etkileyen faktörlerin incelendiği bir çalışmada, splenomegali ve splenektominin kemik iliği hastalığı riskini arttırdığı görülmüştür. N370S allelinin
kopya sayısı fazla olan hastalarda ise kemik iliği hastalığı yükününün belirgin olarak daha az olduğu gösterilmiştir 77.
Osteosarkomlar veya osteoblastomlar dahil olmak üzere sekonder kemik tümörleri çok nadir olarak bildirilmiştir 37.
Kemik belirtilerinin patofizyolojisi çok iyi bilinmemekle birlikte, ağrılı kemik krizi muhtemelen iskemik vasookluzif olaylarla bağlanır. Kemik belirtileri reverzible olabilirler ve tıbbi görüntülemede lezyon olarak görülmezler. Bununla birlikte, genellikle uzun kemikler (metafizler veya diyafizler) ve yassı kemiklerde kemik infarktları, epifizlerde avasküler nekroza (AVN) neden olurlar 20
.
İskemik olayları (kemik enfarktüsü ve AVN) açıklayan damar teorisine ilaveten, özellikle femur başı, femur kondili ve tibia platosunda spontan veya trabeküler mikrokırığı (mekanik veya spontan AVN) açıklamak için mekanik bir teori öne sürülmüştür. Patofizyolojik mekanizma, kemik iliği veya immün hücrelerdeki değişiklikler, iltihaplanma, makrofaj kaynaklı faktörler, sitokinler ve hormonlar gibi diğer potansiyel faktörleri de içerebilir 20
. Görüntüleme çalışmaları, GH ve/veya komplikasyonlarının belirlenmesinde çok büyük öneme sahiptir. X-ray uzun kemik incelenmeleri (en iyi proksimal tibia ve distal femur grafisinde), trabeküler yapı kaybı, kortikal kemik veya büyük kistik alanların belirlenmesinde kullanılır. Bunlara ek olarak femur başı kırıkları, kemik infarktları ve avasküler nekroz da X-ray`le görülebilir. Metafizin anormal modellenmesinden kaynaklanan distal femurun (dar boyunlu ve düz alt tabanlı konik bir laboratuar şişesi) Erlenmeyer flask deformitesi GH'li hastaların yarısında vardır 39
. Bununla birlikte, bu deformite Gaucher hastalığı için spesifik değildir ve fibröz displazi, Niemann-Pick hastalığı, osteopetroz, ağır metal zehirlenmesinde de görülmektedir.
Şekil 2.2. Erlenmeyer Flask deformitesinin röntgen filmi, kortikal inceltme ile birlikte konik bir şişeye benzeyen dia-metafiz birleşiminde eğri eksikliğini gösterir (sağdaki resim) 78
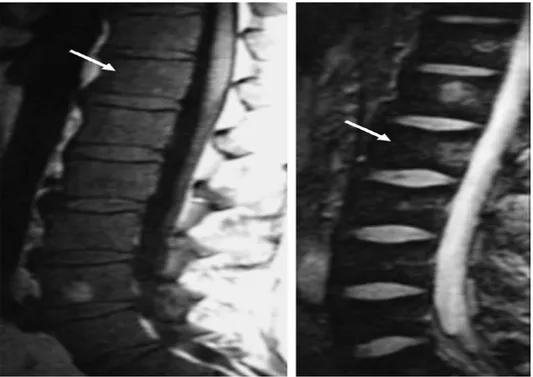
Kemik tutulumu, omurga ve femurun MRG taramalarında en iyi şekilde değerlendirilir. T1 ağırlıklı MRG, "sarı kemik iliği" olarak görülen kemik iliği infiltrasyonunun değerlendirilmesinde kullanılır. T2 ağırlıklı MRG, aktif kemik infarktlarının saptanmasında en hassas yöntemdir .
Şekil 2.3. Lomber vertebranın T1 ve T2 görüntüleri, vertebra cisimlerinde osteoskleroz değişikliklerini temsil eden kemik iliğinde (oklar) bir azalmayı ortaya koymaktadır 78
Lomber vertebra ve femurun dual enerji X-ray absorbsiyometri (DEXA), Kemik mineral dansitesini (KMD) ölçmek için kullanılan standart yöntemdir 79.
Gaucher hastası çocuklarda büyüme geriliği sıklıkla gözlenir. Genellikle doğumda ve erken çocukluk döneminde boy persentilleri normal sınırlar içerisindedir, ancak ilerleyen çocukluk döneminde büyüme hızı azalmakta ve bu nedenle Gaucher hastaları boy açısından yaşıtlarının gerisinde kalabilmektedir. Bu hastaların tedavi ile nihai olarak normal boya ulaşabildiği gösterilmiştir. Aynı zamanda, GH çocukların, puberteye yaşıtlarından daha geç girdiği ve bu dönemi daha geç tamamladığı bilinmektedir. Bu anormalliklerin temelinde kronik anemi ve hepatosplenomegali ile birlikte istirahatte harcanan enerjinin de artmış olması da yatmaktadır 80
.
2.8.4 Akciğer Tutulumu
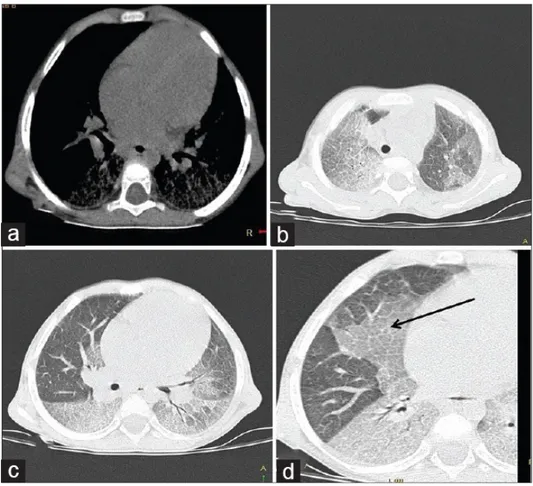
Akciğer tutulumu tüm GH tiplerinde görülebilmekte ve daha çok şiddetli klinik seyirle ilişkilidir. Klinik olarak asemptomatik ve şiddetli solunum yolu semptomları, normal veya hafif radyografik değişikliklerden belirgin radyografik bulgulara kadar değişen geniş bir yelpazede kendini gösterir. Akciğer tutulumu, Gaucher hücreleri tarafından alveol, interstisyum, bronş veya pulmoner damarların infiltrasyonunun bir sonucudur 81. İnterstisyel ve alveolar tutulumun kombinasyonu akciğer tutulumunun nadir tipidir, Arat A. ve ark. 82tarafından yapılan bir olgu sunumunda tanımlanmıştır.
Akciğer tutulumu Tip 2 ve 3'de (şiddetli formlar) görülür. Hepatopulmoner sendrom nedeniyle AV şant`lar görülmektedir 81
. GH`da pulmoner arteriyovenöz şantlar ile birlikte ekzoftalmus veya talasemi hastalarına benzer yüz görünümünün olduğu farklı fenotipik özellikler de tanımlanmıştır 83. Hepatosplenomegali, restriktif akciğer hastalığı, veya kemik tutulumuna bağlı kifozun ekstrinsik bası ile akciğer fonksiyonunu bozması sonucu oluşabilir. Organomegaliden dolayı diyafragmatik hareketlerin kısıtlanması da pulmoner semptomlarıın artmasına neden olur. Pulmoner hipertansiyona bağlı olarak egzersize bağlı dispne, senkop, ilerleyen dönemlerde istirahat dispnesi, siyanoz ve parmaklarda çomaklaşma olur. Yetişkinlerde, splenektomi ve bazı genetik faktörlerin (N370S mutasyonu dışında bir mutasyona ve pozitif aile hikayesine sahip olmak, ACE I gen polimorfizmi ve kadın cinsiyeti) varlığında ciddi pulmoner hipertansiyon riskinin pediatrik papulyasyona göre daha
fazla olduğu bildirilmiştir. GH`da L444P homozigot mutasyon olan hastalarda pulmoner tutulum riski daha da yüksektir 84.
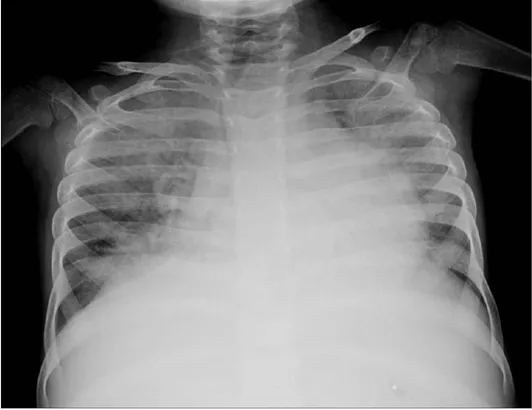
Solunum şikayetleri ile gelen hastalarda göğüs röntgen ve HRCT yapılabilir. Göğüs röntgen filmi normal olabilir veya diffüz retiküler tutulumu gösterir. HRCT 'de akciğer tutulumunun üç farklı örneği görülebilir yani sadece interstisyel tutulum veya sadece buzlu cam görünümü veya ikisinin kombinasyonu olabilir 81.
Şekil 2.4. Bilateral akciğer alanlarında retikülonodüler işaretleri gösteren göğüs radyografisi PA görünümü 83
Şekil 2.5. (a-d) HRCT akciğer penceresi aksiyel görüntüler. (b-d) Buzlu cam üst loblar (b) ve bazlar seviyesinde interlobüler ve intralobüler septal kalınlaşma 83.
Gaucher hastalığında pulmoner hipertansiyon yaygın değildir, ERT uygulanması sonrası pulmoner hipertansiyonda azalma mevcuttur. Bununla birlikte, ERT alan erişkin ve çocuklar da dahil olmak üzere, hastalarda pulmoner hipertansiyonu zamanında yakalamak için, ekokardiyografi, erken teşhis için önerilmektedir. Triküspid yetmezlik (TY) gradiyent analiziyle zirve pulmoner arter basıncı tahmin edilir. TY gradiyenti için normalin üst sınırı 30 mm Hg kabul edilmektedir. Kontrastlı ekokardiyografi ile intrapulmoner şantlar gösterile-bilmektedir. Literatürde %45 kadar yüksek oranlarda solunum fonksiyon testi (SFT) anormallikleri bildirilmiştir. Temel incelemeler içindegörüntüleme tetkikleri, (SFT) ve ekokardiyografi olmalıdır 85
.
2.8.5. Nörolojik Tutulum
GH tipleri primer santral sinir sistemi (SSS) tutulumunun olup olmamasına bağılı olarak üç alt tipe ayrılır. Akut tip 2 ve subakut tip 3 gaucher hastalarında
nörolojik tutulum (örn., Nöbetler ve bilişsel gerileme) mevcuttur. GH tip1`de radikülopati veya omurilik kompresyonundan kaynaklanan nörolojik bulgular öncelikli olarak ikincil komplikasyonlardır.
Tip 2 GH'li hastalarda, iki yaştan sonra hızla ilerleyen nörolojik semptomlar, sınırlı psikomotor gelişim ve semptomların başlangıcından sonrakı 2 yılda ölüm görülmektedir. Ciddi şekilde etkilenen hastalardan oluşan bir alt grupta kolodion cilt değişiklikleri veya non-immün hidrops fetalis ile yaşamın ilk birkaç haftasında ölüme neden olabilir 4. Tip 3 GH'li hastalarda, semptomlar iki yaşından önce başlamış olabilir, ancak bazı durumlarda genellikle ömrün üçüncü veya dördüncü on yılına uzayan daha yavaş ilerleyici bir seyre sahiptir. Tip 2 GH'li hastalarda öngörülebilir bir seyir izlerken, tip 3 GD olanlar hafif veya şiddetli viseral tutulum ile ilişkili olabilen anormal horizontal sakkadlarla heterojen bir prezentasyona sahip olabilirler. Bu hastaların bazılarında genel olarak tonik-klonik nöbetler ve ilerleyici miyoklonik epilepsi görülmüştür 86. Nöronopatik gaucher hastalarında (NGH), beyin sapı işitsel uyarılmış yanıt (BAER) testi beynin anormal dalga formları ve manyetik rezonans görüntüleme (MRI) hafif serebral atrofi gösterebilir 52.
Tip 1 GD'li hastalarla ilgili son çalışmalar, periferik nöropati veya parkinsonizmin daha önce belirtilmiş olanlardan daha yaygın olabileceğini göstermektedir. İlginç bir şekilde, aile çalışmaları parkinsonizme ilişkin riskin, yalnızca etkilenen kişiler (iki mutant alel) için değil, aynı zamanda taşıyıcılar için (bir mutant alel) de arttığını göstermektedir 87.
2.8.6. Hiperimmünoglobulinemi ve Maligniteler
GH`da hiperimmünoglobulinemi sık karşılaşılan bir bulgudur. Bu bulgu, makrofajlarda depolanan glikoserebrozidin immun sisteme kronik uyarısıyla, plazma hücreleri tarafından aşırı miktarda immünoglobulin üretiminden kaynaklanmaktadır. Yakın dönemdeki çalışmalar ile Gaucher hücrelerinin proinflamatuvar sitokin salgılaması sonucu B hücre serisinin stimulasyonu olabileceği ve hiper-immünoglobulinemi ile klonal ekspansiyona, ardından da B hücre malignitelerine yol açabileceği gösterilmiştir 88, 89
.
Gaucher hastalığında multipl miyelom (MM) insidansı artmaktadır. Dünya genelinde 2742 Gaucher hastası olan Uluslararası Gaucher Kayıt sisteminde yapılan incelemede, MM insidansının 5 - 9 kat arttığını ortaya koymuştur Gaucher

![Tablo 3.1. Olgu formu: Gaucher hastalığı izlemi [ tüm yaş grupları ve tüm tedaviler (ERT, Substrat redüksiyon) ]](https://thumb-eu.123doks.com/thumbv2/9libnet/3036117.2626/50.892.150.817.162.1155/tablo-gaucher-hastaligi-izlemi-gruplari-tedaviler-substrat-reduksiyon.webp)