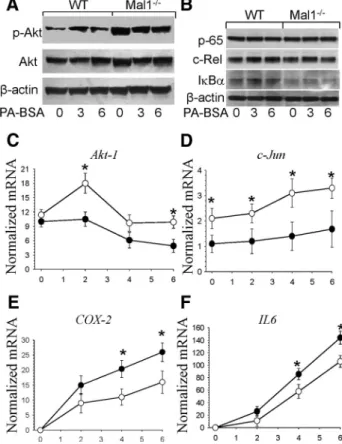
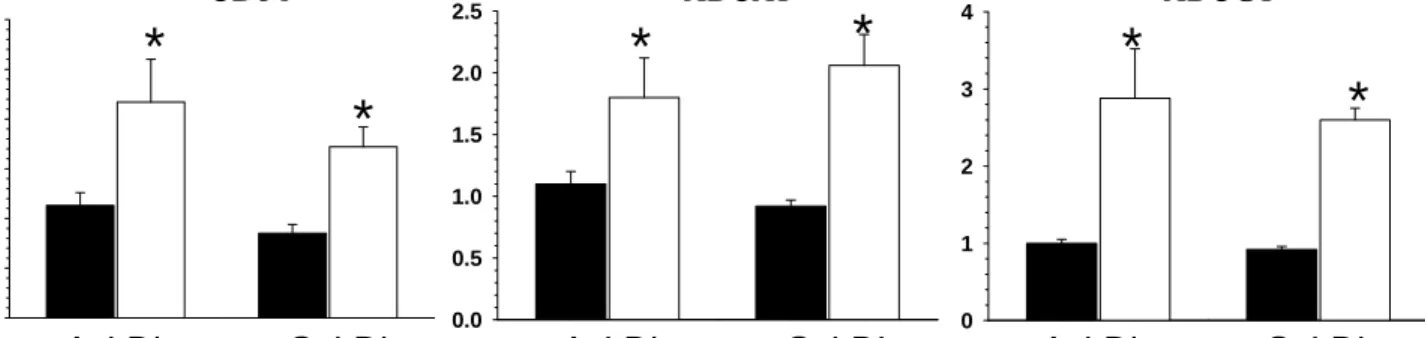
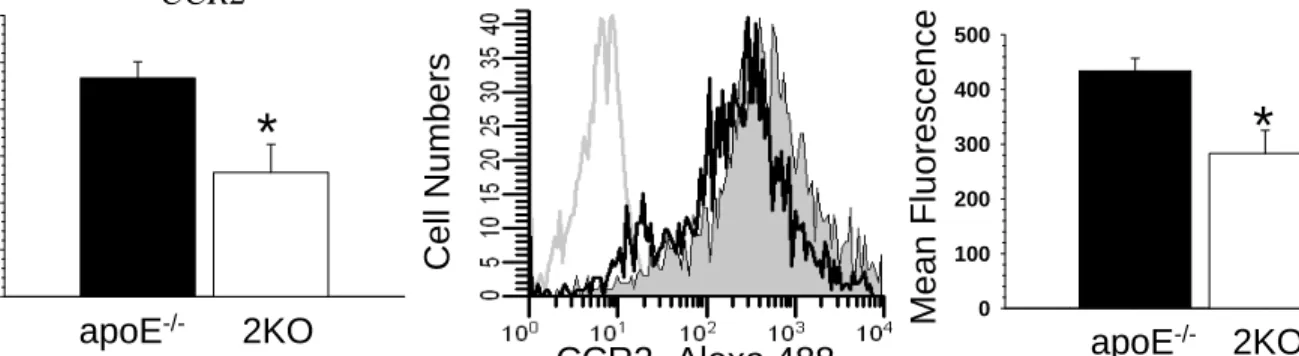
Macrophage Mal1 deficiency suppresses atherosclerosis in low-density lipoprotein receptor-null mice by activating peroxisome proliferator-activated receptor-g-regulated genes
Tam metin
Şekil
Benzer Belgeler
These results indicate that the activated Notch1 receptor and -enolase or MBP-1 cooperate in controlling c-myc expression through binding the YY1 response element of the
This study examined the association between pressure pain sensitivity and various single nucleotide polymorphisms (SNPs) of human μ -, κ -, and δ -opioid receptor (i.e. OPRM1,
Sigortahmn i~yerinde bulundugu srrada, i~veren tarafmdan garev i l e ba~ka bir yere gan- derilmesi ytiztinden as Il i~ini yapmakslZln ge~en zamanlarda, emzikli kadm
Because the cellular mechanism of absence seizures indicates the involvement of ion channels in the pathogenesis of absence epilepsies; gene analysis carried out both on patients and
In the decision-level fusion scheme with global (sensor-independent) training model, a common classification model is used for the feature vectors extracted from
In situ photoinduced crosslinking of the intercalated monomer and the PSU macromonomer in the silicate layers leads to nanocomposites that are formed by individually
Previously our group showed that oval cell marker, FLT3 changes its cellular localization in response to partial hepatectomy and suggested a role in liver regeneration.. In
IL-6 (-572G/C rs1800796) and IL-6R (1:G.154448302 T > C rs7529229) gene polymorphisms may have an impact on cytokine production, immune response and these gene polymorphisms may
