Characterizing microsatellite polymorphisms using assembly-based and mapping-based tools
Tam metin
Şekil
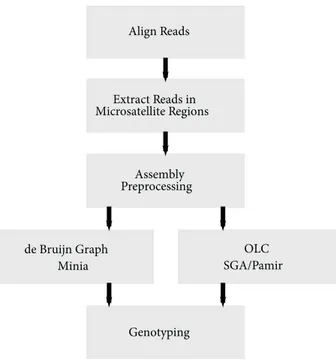
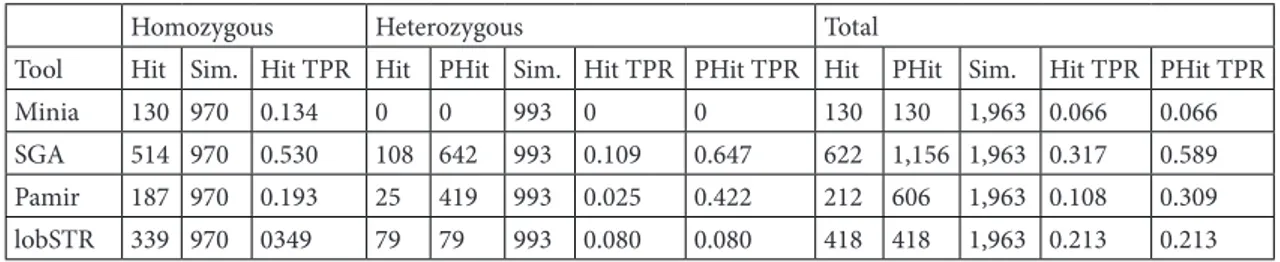

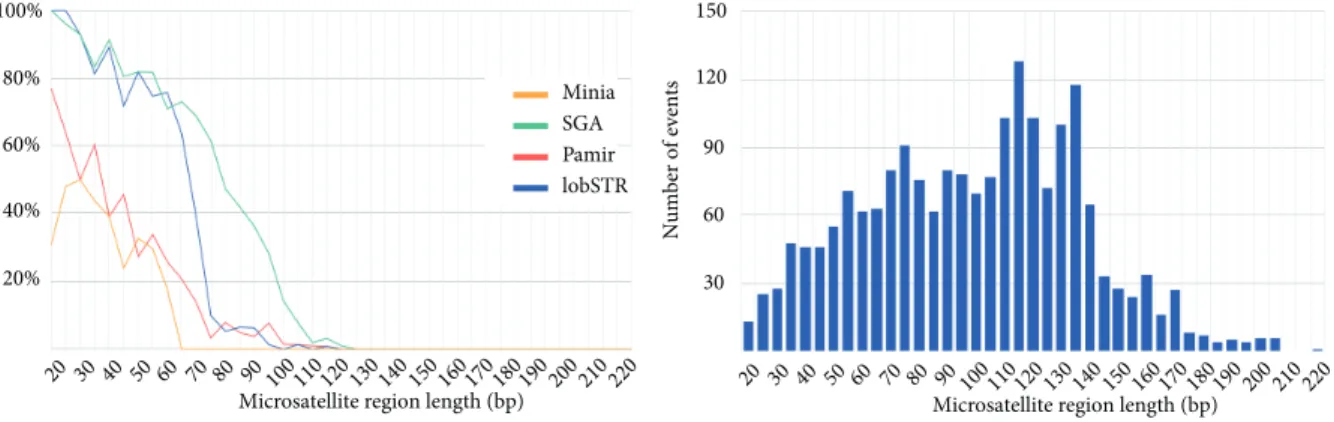
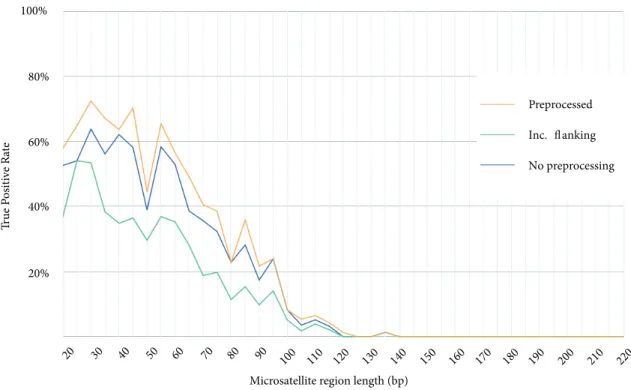
Benzer Belgeler
He firmly believed t h a t unless European education is not attached with traditional education, the overall aims and objectives of education will be incomplete.. In Sir
In- terestingly, from the QMC correlation energy calculations we find that under an externally applied magnetic field the 2D electron system undergoes a first-order phase transition
While automatically selecting the suitable cues and rendering methods for the given scene, we consider the following factors: the distance of the objects in the scene, the user’s
Consistent with our two postulates, we suggested 1 - 3 defining the fractional Fourier transform as the change of the field caused by propagation along a quadratic
Sonuç olarak, atorvastatinin intimal hiperplazi üzerine etkisi plazma kolesterol düzeyinden bağımsız olarak düz kas hücresi, endotel ve diğer vasküler
MPLS automatic bandwidth allocation (or provisioning) refers to the process of dynami- cally updating the bandwidth allocation of a label switched path on the basis of actual
A total of 200 measurements were performed (10 patients×16–22 measurements per patient). We found that for all realistic bilateral lead trajectories, there existed an optimum
Bu çalışmada ebeveynlerin çocuklarının bilgisayar kullanımını ve internet erişimini kontrol ve takip edebilmelerine olanak sağlayan, kullanımı kolay olan, açık
