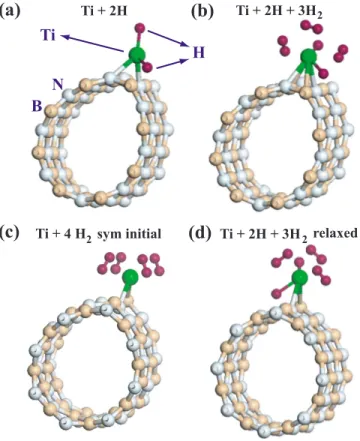
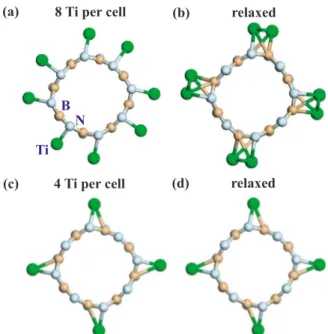
Hydrogen storage capacity of Ti-doped boron-nitride and B Be-substituted carbon nanotubes
Tam metin
Şekil
Benzer Belgeler
Thyrotoxicosis began to improve, and the patient was discharged with ampicillin-sulbactam (750 mg) given orally twice daily, and antithyroid therapy on the 14 th day of
Moreover, with the appropriate choice of excess and shortage costs incurred at the end of a single period, it also provides a good myopic approximation for an infi- nite
The pro- posed approach applies progressive morphological filtering to compute a normalized DSM from the LiDAR data, uses thresholding of the DSM and spectral data for a
We model the problem as a two stage stochastic mixed integer nonlinear program where the first stage determines the departure time of new flights and the aircraft that is leased..
As the operation is done in liquid environment, the radiation impedance has to be modeled correctly and included in the mechanical side of the circuit model. It is important to
In this study, composite films of PEK and PPy were obtained by electropo- lymerizing pyrrole in a PEK substrate coated on a platinum electrode in an electrolyte solution of
FIR süzgeç tasarım problemi öncelikle dı¸sbükey olmayan karesel kısıtlamalı karesel programlama (QCQP: Quadratically Constrained Quadratic Pro- gramming) olarak modellenmi¸s,
(Color online) Emission spectra of our NC integrated white LED achieving a high S/P ratio of 3.05 at a CRI of 71.0 under various current injection levels at room temperature, along
