T.C.
EGE ÜNİVERSİTESİ TIP FAKÜLTESİ
ÇOCUK SAĞLIĞI ve HASTALIKLARI ANABİLİM DALI Prof. Dr. Savaş KANSOY
LANGERHANS HÜCRELİ HİSTİOSİTOZ’LU
OLGULARDA SANTRAL SİNİR SİSTEMİ
HASTALIĞININ ARAŞTIRILMASI
UZMANLIK TEZİ
Dr. Hamiyet HEKİMCİ ÖZDEMİR
Tez Danışmanı: Prof. Dr. Nazan ÇETİNGÜL
Tüm uzmanlık eğitim sürecim içinde bilgi ve tecrübelerini benden esirgemeyen, başta kliniğimiz Anabilim Dalı Başkanı Prof. Dr. Savaş Kansoy ve tüm hocalarıma teşekkürediyorum.
Tıp fakültesi eğitim sürecimden bu yana, hayatın her alanında, her zaman danışabildiğim bir hoca , manevi desteğini hep hissettiğim bir büyüğüm olan, çok iyi bir hekim olabilmek için kendisini örnek aldığım Prof.Dr. Nazan Çetingül’e sonsuz teşekkürler.
Pediatri uzmanlığını tercih etmeme neden olan, çocuk sevgisi ve çalışma azmini bana kazandıran, bu çalışma sırasında her türlü yardımlarını esirgemeyen, çocuk onkoloji bölümü hocalarım, Prof. Dr. Savaş Kansoy, Prof.Dr. Nazan Çetingül, Prof. Dr. Mehmet Kantar, Prof. Dr. Serap Aksoylar , her zaman desteğini hissettiğim Kamile Abla ve tüm bölüm çalışanlarına teşekkür ediyorum.
Çalışmada, hastaların nörolojik bakılarının yapılması, değerlendirilmesi, EEG’lerinin çekilerek yorumlanması aşamalarında yardımlarından dolayı , Prof. Dr. Sarenur Gökben, Prof. Dr. Gül Serdaroğlu, Uzm. Dr. Sanem Keskin Yılmaz, Uzm. Dr. Hande Gazeteci Tekin, Ped. Nöroloji BD EEG laboratuarı ve poliklinik çalışanları Ayla Savaş, Gizem Kızıldağ, Erdem Turan, Vildan Yıldız ‘a çok teşekkürler.
Hastaların kraniyal MRG çekimi ve yorumlanması aşamasında yardımlarından dolayı Prof. Dr. Cem Çallı, Prof. Dr. Ömer Kitiş ve 3 Tesla MR çalışanlarına çok teşekkürler.
Zorlu hayat yolunun en büyük adımı olan pediatri asistanlık sürecinde, sakinliğini ve sabrını hiç eksik etmeyen, huzurum, hayat arkadaşım, sevgili eşim Dr Taha Reşid Özdemir ‘e varlığından ötürü sonsuz teşekkürler.
Dr. Hamiyet HEKİMCİ ÖZDEMİR İzmir,2013
ÖZET
LANGERHANS HÜCRELİ HİSTİOSİTOZ’LU OLGULARDA SANTRAL SİNİR SİSTEMİ HASTALIĞININ ARAŞTIRILMASI
Langerhans hücreli histiositoz (LHH), dendritik hücre sistemine ait CD1 a pozitif Langerhans hücreleriyle beraber lenfosit, normal histiosit/makrofajların proliferasyonu sonucu doku ve organlarda birikimi ile karakterli ve sıklıkla çocukluk çağında görülen nadir bir hastalıktır. Hastalık yaygınlığı tek kemik lezyonundan- multisistem organ yetmezliğine kadar değişebilmektedir. Hastalık seyri spontan remisyondan, yıllar içinde kronik rekürrenslere ve ölümcül organ yetmezlik bulgularına değişkenlik gösterebilir. LHH ‘da SSS hastalığı nadir olmakla beraber, morbiditesi yüksek oluşuyla önem taşımaktadır.Küçük yaş, MS hastalık tutulumu ve kraniyofasiyal kemik tutulumlarının risk oluşturduğu SSS hastalığında Dİ en sık rastlanan hastalık bulgusu olarak bildirilmektedir. Geçen 20 yılda SSS hastalığında semptomatik ve asemtomatik olgularda çeşitli kraniyal MRG bulguları tanımlanmıştır.
Çalışmada birincil amacımız multidispliner yaklaşımla LHH’li olgularda SSS hastalığının nöropsikiyatrik bakıları ve kraniyal MRG değerlerlendirmeleri ile belirti ve bulguları saptayarak, SSS hastalığının sıklığını belirlemektedir. Ikincil olarak SSS hastalığını saptamada önemli yer tutan kraniyal MRG yöntemindeki özellikleri belirlemek, standardize etmek ve olguların tanı ve izlemlerinde radyolojik incelemenin önemini vurgulamak amaçlanmıştır.
Bu çalışma Ege Üniversitesi Tıp Fakültesi Çocuk Sağlığı ve Hastalıkları A.D. Çocuk Onkoloji Bölümünde Langerhans Hücreli Histiositoz tanısı ile izlenen 30 olguda SSS hastalığının sıklığını ve özelliklerini belirlemek üzere planlanan kesitsel geriye dönük özellikte bir çalışmadır. ( Etik kurul numarası: No1396/596, BAP proje no:2013-TIP-90)
Hastaların geçmiş izlem bulguları dosya taraması ile belirlendi. Çalışma kapsamında nörolojik bakıları EEG, kraniyal MRG çekimleri çocuk nörolojisi bilim dalı ve radyoloji ABD’ larınca yapıldı.
Çalışmada tanı yaş ortalaması 81,3 ay (8-202 ay) ortanca 74 ay , erkek/ kız oranı 1,1 olarak bulundu. Küçük yaş olarak değerlendirilen 36 ay altı hasta sayısı % 36 (11), bunların %63’ünde ( 7) multisistem tutulum saptandı. Olgularda en sık
tutulan iskelet sistemi (%97) olup, %66 (20) tek sistem tutulumu %34ü (10) multisistem tutulumu gösteriyordu.SSS hastalığı açısından riskli kemik tutulumu olan 7 olgunun ,6’sında temporal kemik, 1’inde orbital kemik tutulumu vardı.
SSS hastalık bulguları olan 10 (%33,3) olgu saptandı. Bunların 6’sı radyolojik ve klinik bulguyu birlikte gösteren “kanıtlı SSS hastalığı” olarak kabul edildi. 4’ünde radyolojik bulgularına ek, risk organ tutulumu ve/veya gelişim basamaklarında gerilip olması nedeniyle “kuvvetle olası SSS hastalığı” olarak değerlendirildi. Kraniyal MRG’da LHH ile uyumlu olabilecek lezyonları gösteren, SSS hastalığı için risk taşımayan, 7 olgu da “kuşkulu SSS hastalığı?” açısından sıkı izlem gerektiren olgular olarak düşünüldü.
SSS hastalığı olarak kabul edilen 10 olgunun 9’u MS tutulumlu olup , 6’sı 3 yaş altında idi, böylece MS tutulum ve küçük yaşın SS hastalığı açısından risk oluşturduğu görüldü
Riski yüksek hastalarda hastalığa özgü primer sağaltım yanında IVIG uygulamasının bulguları önleyebileceği ve geriletebileceği gözlendi
Sonuçta özellikle küçük yaşta MS tutulum gösteren LHH’li olgular öncelikli olarak, tüm olguların SSS hastalığı riski taşıdığı unutulmamalıdır. Ayrıca multidisipliner olarak, SSS hastalığının saptanmasında, tanı ve izlemde nöropsikiyatrik bakı ve uygun teknikle yapılan kraniyal MRG değerlendirilmesi büyük önem taşımaktadır.
Anahtar Kelimeler: Langerhans hücreli histiositoz, santral sinir sistemi hastalığı, LHH’ lu hastalarda nöroradyolojik görüntüleme
ABSTRACT
INVESTIGATION OF CENTRAL NERVOUS SYSTEM DISEASES IN LANGERHANS CELL HISTIOCYTOSIS
Langerhans cell histiocytosis (LCH) is a rare disease in childhood involving clonal proliferation of CD1a positive Langerhans cells, abnormal cells deriving from bone marrow and capable of migrating from skin to lymph nodes. Clinically, its manifestations range from isolated bone lesions to multiple organ failure. Progression of the disease may vary from spontaneous remission to chronic relaps and fatal multiple organ failure over the years. CNS-LCH is a rare significant form of LCH due to its high morbidity rate. Diabetes insipidus is most commonly caused in CNS-LCH disease in which early age, craniofacial bone and MS disease involvements constitute a risk. In the past twenty years, several cranial MRI findings of symptomatic and asymptomatic cases have been identified in CNS-LCH disease. The first aim of our study was to determine, via a multidisciplinary approach, the frequency the frequency of CNS disorders at LCH cases using the clinical findings obtained from neuropsychiatric exams and cranial MRI evaluations. A second aim was to determine and standardize the features of cranial MRI method, which is very important in detecting the CNS disorders, as well as to emphasize the importance of radiological examination used in the diagnosis and management of patients.
This is a retrospective and cross-sectional study to determine the frequency and characteristic features of CNS disease in 30 LCH patients followed in Ege University, Faculty of Medicine, Department of Pediatric Oncology. (Ethics committee number: No1396/596, Commission project number : 2013-TIP-90) Patient follow-up results of the past were determined by scanning files. In the study, EEG, neurological examination, cranial MRI was done by departmant of pediatric neurology and radiology.
In this study, the average age at diagnosis was 81.3months of age (8-192 months), with median of 74 months of age, and the male to female ratio was found as 1.1. The number of patients under 36 months old, i.e., considered as early age, was 36% (11). In 63% (7) of those, multisystem clinical involvement was detected. Skeletal system was the most commonly affected system (97%). 20 (66%) of all cases showed single-system involvement whereas 10 (34%) of them showed
multisystem involvement. There were temporal and orbital bone involvement in 6 of 7 and 1 of 7 cases respectively who had a risk for bone involvement due to the CNS disease.
Signs of CNS disorders were detected in 10 (33%) cases Six of them have clinical and radiological disease findings approved as “certain CNS disease”. Since four patients have developmental delays and/or the risk of organ involvement in addition to the radiological findings They were approved as as "highly probable CNS disease". There were 7 cases with MRI lesions that are compatible with LCH, but do not have a risk for CNS disease. These cases were approved as “suspected CNS disease?” and follow-ups were scheduled.
Since there were 9 patients with a multisystem disease (out of 10 diagnosed with “certain CNS disease”) and 6 under three years of age, we conclude that multisystem involvement and early age have a higher risk of developing CNS manifestations. It was found that in patients at high risk of disease specific primary treatment in addition IVIG therapy could prevent and reverse the findings.
In conclusion, it should be noted that all cases especially the ones with MS involvement at an early age have a risk in terms of CNS disease. In addition, from a multi-disciplinary perspective, neuropsychiatric exams and cranial MRI scans are crucial in the diagnosis of CNS disease, as well as in the management of patients.
Keywords : Langerhans cell histiocytosis, CNS-LCH disease, neuroimagining of LCH patients
KISALTMA LİSTESİ
LHH : Langerhans Hücreli Histiositoz LH : Langerhans hücresi
BG : Birbeck granulü
SSS-H : Santral sinir sistemi hastalığı Dİ : Diabetes insipitus
DEA : Demir eksikliği anemisi TS-LHH : Tek sistem LHH
MS-LHH : Multisistem LHH (MS-LHH) RO : Riskli organ
RO+ : Riskli organ tutulumu olan RO- : Riskli organ tutulumu olmayan KIT : Kemik iliği transplantasyonu HKHN : Hematopoetik kök hücre nakli ARA-C : Sitozin arabinosid
2-CdA : 2- klorodeoksiadenozin MTX : Metotreksat VBL : Vinblastin PRED : Prednizolon 6-MP : 6 merkaptopürin VCR : Vincristin MMF : Mikofenolat mofetil
EDSS : Genişletilmiş Özürlülük Durum Ölçeği
ICARS : Uluslararası Kooperatif Ataksi Derecelendirme Ölçeği
MABC-2 : Çocuklar için hareket değerlendirme bataryası-2
IVIG : İntravenöz immunoglobulin
AGTE : Ankara gelişim envanteri
1. GİRİŞ
Langerhans hücreli histiositoz (LHH), dendritik hücre sistemine ait CD1 a pozitif Langerhans hücreleriyle beraber lenfosit, normal histiosit/makrofajların proliferasyonu sonucu doku ve organlarda birikimi ile karakterli ve sıklıkla çocukluk çağında görülen nadir bir hastalıktır. Çocuklukta LHH’nin tek sistem tutulumu sık olup, çok odaklı veya multisistem tutulumuna varan farklı klinik özellikler gösterebilir. Hastalar sıklıkla iskelet sistemi tutulum bulgularıyla tanı almakta ve bu grupta sağaltıma yanıtları iyi olmaktadır. Küçük yaş grubunda daha sık görülen multisistem tutulumda, tutulan organ disfonksiyonuna bağlı olarak, hastalık progresif seyir gösterir. İskelet sistemi, karaciğer cilt, kemik iliği, SSS belli başlı hastalığın bulgularının olduğu organ ve sistemlerdir. Spontan düzelen hastalar olabildiği gibi, LHH sağaltımı, hastalığın evresine, lezyon yeri ve yaygınlığına gore local sağaltım, radyoterapi, çeşitli kemoterapi uygulamaları yanında multisystem hastalıkta kök hücre nakli gerektirebilir.
Tanı, tutulan doku veya organlardan alınan biyopside CD1a ve S100 pozitif hücrelerin gösterilmesi ile konur.
Hastalığın en sık tutulumu kemik (%80) olup, SSS hastalığı multisistem tutulumlu olguların %10-20 sinde saptanır. Sıklıkla hipotalamo-hipofiz tutulumu sonucu gelişen diabetes insipitus (Dİ; multisitem hastalıkta veya risk lezyon varlığına gore %5-40 arasında görülmekte) endokrinopatiler, hipotalamik disfonksiyon, nörodejeneratif bozukluk veya nadiren yer kaplayan lezyonlar şeklindedir. Hastaların sağaltımı ile kür sağlansa bile Dİ kalıcıdır. LHH’de SSS hastalığının önemine son 10 yılda dikkat çekilmektedir. SSS lezyonlarının ortanca 3 yılda geliştiği, nörolojik bulguların bazı olgularda 20 yıl içinde ortaya çıktığı belirtilmektedir.
Izole SSS tutulumu saptanan ve/ veya diğer system tutulumlarına SSS tutulumu eşlik ettiği hasta grubunda, radyolojik tanı önemli yer tutmaktadır. LHH nin SSS tutulumunda radyolojik tanı kriterleri belirlenmiştir. Temporal ve orbital kemik veya kafa kemikleri tutulumlu multisitem hastalıkta SSS hastalığının riski fazladır. Klinik bulgular (ataksi, nöbet geçirme, Dİ, endokrinopati gibi) olsun veya olmasın kraniyal- MRG çekilen hastalarda, LHH hastalığına ait bulgular saptanabilir. Nöropatolojik bulguların ortanca 3 yıl içinde geliştiği, bu sürenin 20 yıl kadar uzayabildiğini gösteren çalışmalar dikkat çekicidir. Hastaların çoğunda farklı SSS bulgularının
kombinasyonu görülebilir. LHH-SSS hastalığının tanınması, sonraki izlem ve gerekli sağaltımın planlanması açısından büyük önem taşımaktadır.
Bu çalışmada birincil amacımız multidispliner yaklaşımla LHH’li olgularda SSS hastalığının nöropsikiyatrik bakıları ve kraniyal MRG değerlerlendirmeleri ile belirti ve bulguları saptayarak, SSS hastalığının sıklığını belirlemektedir. Ikincil olarak SSS hastalığını saptamada önemli yer tutan kraniyal MRG yöntemindeki özellikleri belirlemek, standardize etmek ve olguların tanı ve izlemlerinde radyolojik incelemenin önemini vurgulamaktır.
2. GENEL BİLGİLER
2.1 Histiositoz sendromları
Histiositozlarla ilk olgu 1893 yılında Alfred Hand tarafından bildirilen poliüri, ekzoftalmus, hepatosplenomegali ve deri döküntüleri olan 3 yaşında bir olgu olup, otopsi materyalinde granulamatöz lezyonları belirtilmiştir. 1920’li yıllarda benzer olguların saptanmasıyla hastalığın klasik triadı; poliüri, eksoftalmus ve kemik defektleri olarak tanımlanmış, Hand-Schüller-Christian hastalığı adıyla literatürde yerini almıştır.(1)
1924 yılında Erich Letterer’in tanımladığı 6 aylık olguda, multisistemik tutulum mevcuttur. Olgunun otopsisinde retikulum hücre proliferasyonu meydana gelen, birçok organ yerleşimli iri hücreler saptanmıştır. 1933 yılına gelindiğinde Sture Siwe de benzer özellikte olgular saptamıştır. Böylece sistemik retiküloendotelyoz Letterer-Siwe hastalığı olarak tanımlanmıştır.(2,3) Son histiositoz sınıflaması 1997 yılında tanımlanmıştır (Tablo 1). (4)
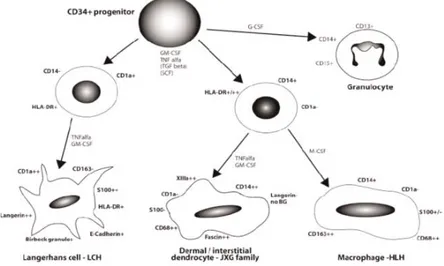
Histiosit, makrofajların dokularda olan adıdır. Makrofajlar ve dendritik hücreler aynı kökene sahiptir ve CD34+ hücreden meydana gelirler. Gelişimleri sırasında karşılaştıkları sitokinler ile yerleştikleri bölgelere göre Langerhans hücresi, dermal/intertisyal dendrosit veya makrofaja dönüşebilirler. (5,6)
Histiositoz ile ilişkili hastalıkların patogenezinde rol oynayan immun hücre grubunu mononukleer fagositer hücreler ve dentritik hücreler oluşturmaktadır. Makrofajlarrın doku tamiri dışında tumör baskılamak, antijen işlemek, immun stimulasyon gibi bir çok görevleri mevcuttur. Dentritik hücrelerin primer görevi ise antijen işlemek ve immun sistem regulasyonudur.(7)
Şekil 1: Histisitositik hücrelerin immun fenotipik işaretleri Tablo 1. Çocukluk çağı histiositik hastalıklarının sınıflaması
2.2.Langerhans Hücreli Histiositoz
Langerhans hücreli histiositoz (LHH), CD1a, langerin ve S100 proteini eksprese eden ve ultrastriktürel incelemede Birbeck granülleri varlığı gösterilen Langerhans tipi hücrelerin dokuda klonal neoplastik proliferasyonudur.
Langerhans hücreli histiositozda görülen hücreler, deride görülen Langerhans hücrelerine göre daha immatür özelliklere sahiptir. Fizyolojik koşullarda Langerhans hücreleri (LH)T lenfositlere antijen sunumu yapar. Hastalık halinde ise tutulum olan organa giderek (kemik, akciğer, karaciğer..) ve T Hücreleriyle beraber sitokin fırtınası yaparak granulom oluşumuna neden olur. (8) Yapılan deneysel gen expresyon çalışmaları epidermal kökenli ve LHH kökenli langerhans hücrelerinin aynı özellikte olmadığını vurgulamaktadır.(9)
Langerhans hücreli histiositoz, nadir olmakla birlikte, erişkin ve çocukluklarda histiositik hastalıkların en sık görülen formudur. İlk tanımlanmasından bu yana geçen yüzyılda, anlaşılması zor bir hastalık grubu olup, tarihte farklı şekillerde isimlendirilmiştir. Geçmişten bugüne LHH, Histiositoz X, eosinofilik granuloım, Abt-Letter-Siwe hastalığı, HanSchuller-Christian hastalığı veya diffüz retikuloendoteliazis, gibi farklı terminoloji ile isimlendirilmiştir.(19) Langerhans hücrelerinin hastalığın patogenezindeki temel rolünün anlaşılmasından sonra, 1985 yılından itibaren LHH olarak adlandırılmıştır. (10)
2.2.1.Epidemiyoloji
LHH, neonatal dönem de dahil olmak üzere her yaş grubunda görülebilir. Literatürde 35 haftalık doğum öyküsü olan bir olguda konjenital cilt tutulumlu LHH tanımlanmıştır. (11) En sık olduğu yaşlar 1-3 yaş arasında olup, erkeklerde daha sık rastlanmaktadır. Tüm dünyada hastalık prevelansı 1/50.000, yılık insidansı 1,08/200,000 olarak bulunmuştur. (7) Son olarak 1973-2010 yılları arasında Amerika’da yapılan 828 LHH’lu olguyu da içeren bir insidans araştırmasında yıllık insidans 0.142/100,000 olarak belirtilmektedir.(6) Sınıf I ve sınıf III histiositozlu hastaların birlikte değerlendirildiği bu çalışmada, tedavi yöntemlerinin etkinliği ve sağ kalım oranları da belirtilmiştir. Bu çalışmaya göre kız ve erkek oranları arasında fark saptanmamış, tanı yaşı sıklıkla (%33 lük bir oranla) 1-4 yaş arasında bulunmuştur. Son 30 yıl geneline bakıldığında, olguların yarısından fazlasının son 10
yılda tanı almış olduğu görülmüştür. Son dönem tedavi yöntemleriyle sağ kalım oranlarının da değerlendirildiği bu çalışmada, her iki grup hastalık için de 5 yıllık sağ kalım oranlarının son 15 yıl içinde belirgin artış gösterdiği görülmüştür. (6)
2.2.2.Etiyoloji ve Patagonez
LHH için kesin genetik bir predispozan faktör tanımlanmamış olmakla birlikte, bazı ailelerde LHH’nın sık görülmesi, monozigot ikizlerde hastalık sıklığının normal popülasyona göre daha fazla olması, LHH’da genetik yatkınlığının olabileceğini düşündürmektedir.(12) Son dönem yapılan bazı genetik çalışmalarda serin-treonin kinaz protein ilişkili (BRAF) mutasyonların LHH ile ilişkili olabileceğini vurgulamakta ve ayrıntılı çalışmalar son dönemde hız kazanarak devam etmektedir.(14,15,16,17)
LHH’dan sorumlu immun mekanizma hala net olarak anlaşılamamıştır. LH veya dentritik hücreler, epidermis, solunum ve genital sistem epidelinde yer alan, myeloid ve lenfoid kökenli antijen sunan hücreledir. Son dönem insan ve farelerde yapılan çalışmalarda da LH’un kemik iliği kökenli monosit prekürsörü saptanamamıştır.(8-10)
Hastalığın gelişiminde, defektif çalışan immun sistem, enfeksiyonlar, çevresel faktörler, hayatın ilk ayların antibiyotik kullanımı, ailede tiroid hastalığı, ishal, kusma, aşılama eksikliği gibi birçok faktör olabildiği düşünülmekle birlikte, kesin sebebi henüz açıklanamamıştır.(18)
Doku dağılımdaki bozukluk sonucu, normal LH’nin dağılımdan farklı olarak hastalıkta kemikler, cilt, lenf bezleri, karaciğer, akciğer, dalak, santral sinir sistemi (SSS), gastrointestinal sistem ve kemik iliği tutulumu olabilmektedir.
2.2.3.Histopatoloji
Langerhans hücreleri kemik iliği kaynaklı olan, normalde deri ve lenf nodunda yerleşen dendritik hücrelerdir. (1) Son çalışmalarda, LH’nin dolaşımda yer alan plazmosit veya monosit kaynaklı hücreler olabileceği de vurgulanmaktadır (2).
Langerhans hücreleri, myeloid marker olarak CD13, CD33i lökosit marker CD45, adezyon molekülü olarak CD40, CD44, ve E-cadherin eksprese etmektedir. Ayrıca, IL-1, IL-6, tumor necrosis factor-a (TNF-a), colony-stimulating factor
(GM-CSF), interferon-c (IFN-c) ve MHC class I/II molekul reseptörü taşımaktadır. LHH’da tanımlanan CD1a+ hücreler, hastalıkta tanımlanan inflamasyon ve tutulum için gerekli kemokinleri (CCL5/RANTES ve CXCL11/I-TAC) salgılamaktadırlar.
Son dönem LHH etiyolojisinde, kontrolsuz hücre proliferasyonundan daha çok vurgulanan, LH eliminasyon ve regulasyonundan sorumlu regulator T hücre (T-regs) yetersizliği ilişkili hastalığın ortaya çıkabileceği hipotezinden bahsedilmektedir. Bu yönüyle gelecek zamanlarda LHH tedavisinde, T-reg araştırmaları önemli olabileceği görülmektedir. (7,11,12, 13)
LHH gelişiminin, malign özellikli veya immun disregulasyon kaynaklı olma olasılığı hala soru işaretleri taşımaktadır. LHH’un neoplastik hastalık olarak sınıflandırılması tartışmaldır. 1990 ların başındann bu yana monoklonal hücre proliferasyonu ve birikimi olması, neoplastik hastalık lehine olup, son dönem yapılan çalışmalarda buna uyan genomic bozukluk tanımlanmamıştır. LHH’da dentritik hücre füzyonundan sorumlu tutulan IL-17A olup, henüz netlike kazanmamıştır. the Histiocyte Society Grubu 1987 yılında, LHH tanı kriterlerini tanımlamıştır. Klasik LHH histopatolojisi:,electron mikroskopisi ile birbeck granullerinin gösterilmesi veya immunohistokimyasal olarak CD1a+ hücrelerin tanımlanmasıdır.(5) İmmunohistokimyasal olarak langerin (CD207), LH yüzey ve sitoplazmasına spesifik lektinin (ctype) gösterilmesi son dönemde BG gösterilmesinde daha pratik olarak tercih edilmektedir. LHH tanısında, hala altın standart olan uygun kliniği olan hastaların lezyonlarında CD1a ve/veya langerin varlığının gösterilmesidir.(5)
LHH, langerhans hücrelerin, dokuda klonal neoplastik proliferasyonu ile oluşur.(13) İnfiltratif özellikte olan hastalık lezyonları granulamatöz, hastalığın erken döneminde proliferatif nitelikte, zamanla lokal destruksiyon ve nekroza ilerlemektedir. Kesin tanısında, lezyon biyopsi materyalinde birbeck granülleri, S-100proteini, CD1a pozitifliğinin gösterilmesi önemlidir. (7)
2.2.4.Klinik Belirti ve Bulgular
LHH’de en sık iskelet sistemi tutulumu (%70-80) olmakla birlikte, multisistemik özellikte de olabilmektedir. İskelet sistemi dışında cilt, lenf nodu, akciğer, timus, karaciğer, dalak, kemik iliği, santral sinir sistemi diğer hastalık tutulumunun olabildiği organlardır. Hastalığın klinik bulguları ve prognozu tutulum yeri ile ilişkili değişebilmektedir (Tablo 2).
Unifokal LHH’li olgular genellikle büyük çocuklar veya erişkinlerdir ve sıklıkla korteksi aşındıran litik kemik lezyonları ile başvururlar. Diğer bölgelerdeki soliter lezyonlar kitle lezyonları veya büyümüş lenf nodları ile prezente olurlar. Multifokal tek sistem LHH, genellikle küçük çocuklarda görülür ve sıklıkla çevre yumuşak dokuyu da ilgilendiren multipl veya ardışık destrüktif kemik lezyonları ile başvururlar. Kafa kemikleri ve mandibulanın tutulumu sıktır. Kranial tutulumu diabetes insipidus izler. Multisistem tutulumlu LHH’li olgular sıklıkla infantlar olup, ateş, sitopeni, cilt ve kemik lezyonlarına ait bulgular ve hepatosplenomegali ile başvururlar.
2.2.4.1.Kemik Tutulumu
Hastaların % 80’inde kemik tutulumu vardır. Hematopoetik olarak aktif kemiklerde ağrılı kemik lezyonları ve/veya şişlik sık bulgulardır. Radyolojik olarak litik kemik lezyonları (sıklıkla zımba ile yapılmış delik), sınırda sklerozu ve periost reaksiyonu bulunmayan tarzdadır. Nadiren de patolojik fraktür olarak kendini gösterebilir (Resim 1). Sıklıkla hastalık bulgularının görüldüğü kemikler kafatası, pelvis, femur, orbita, diğerleri olarak sıralanabilir. Hastaların % 80’inde kemik tutulumu söz konusudur. Mandibula ve maksilla tutulumlarında diş dökülmeleri, vertebra tutulumunda “vertebra plana”, temporal ve mastoit kemik tutulumunda kronik süpüre otitis media, uzun kemiklerinde extremite fraktürleri olabilmektedir. Ayırıcı tanıda düşünülmesi gereken hastalıklar; osteomiyelit, malin kemik tümörleri, kemik kistleri olmalıdır. Lezyonlar düz grafi ile çok net görülmekle birlikte yardımcı bir tanı yöntemi olarak teknesyum 99 kemik sintigrafisi de kullanılabilir. Somatostatin sintigrafisi veya PET de lezyonları gösterebilir, ancak bu iki yöntemin düz grafilere bir üstünlüğü bulunmamaktadır. Kafatası ve mastoid kemikteki lezyonların iyileşmesi tanıdan sonra en az bir yıl sürmekte, ancak iyileşme süreci on yıla kadar bile uzayabilmektedir . Ancak, radyolojik olarak periferik sklerozun görülmesi iyileşmenin ilk bulgusudur. (25,26)
2.2.4.2.Cilt Tutulumu
Cilt % 30-50 oranda sık olarak tutulum gösterir. Diffüz papüler lezyonlar, seboreik egzema, peteşial- purpurik özelliğide içeren veya, granülamatöz ülseratif ve ksantomatöz lezyonlar şeklinde çok değişken cilt bulguları görülmektedir. Lezyonların çoğu koyu pigmentasyon göstererek iyileşir.
2.2.4.3.Lenf Bezi Tutulumu
LHH’da çok sık görülmemektedir. Genellikle multisistem tutulumu olan hastalarda görülür. Timik tutulumda BT’de kalsifikasyon görülmesi tipik bir bulgudur.
2.2.4.4.Akciğer Tutulumu
Tanıda % 10-15 akciğer tutulumu eşlik eder. Tutulum pulmoner disfonksiyon ile sonuçlanabilir. Takipne, dispne, siyanoz, öksürük, pnömotoraks, plevral effüzyon gibi bulgular görülebilir. Radyolojik (grafi- BT) olarak (buzlu cam görünümü) infiltratif, persistan diffüz kistik değişiklikler, noduler infiltrasyonlar, fibrozis ve milier tutulum olarak görülebilir. Tek tutulan sistem olarak düşünüldüğünde kesin tanı akçiğer biyopsisi ile konur.
2.2.4.5. Karaciğer – Safra yolları ve Dalak Tutulumu
Hepatomegali yanısıra hepatik yetemezlik bulgularıyla; hipoproteinemi (<5,5 gr/dl,) hipoalbuminemi (< 2,5 gr/dl), hiperbilurubinemi (> 1,5 mg/dl), ödem, asit ile kendini gösterebilir. Sklorezan kolanjit, kronik karaciğer hasarı olarak düşünülür, f ibrozis, hepatik yetmezlik tablosuna neden olabilir.
Sıklıkla değişken boyutlarda dalak büyüklüğü görülebilir. Hipersplenizmin- trombositopeni nedeni olmaktadır.
2.2.4.6. Hematopoetik Sistem Tutulum
Multisistem tutulumlu LHH’lu olgularda % 50-60 oranda görülür. Bulgular, primer kemik iliği tutulumu ya da hipersplenizme bağlı olarak görülebilir. Kemik iliği yetmezlik bulguları (anemi; < 10 gr/dl, lökopeni, nötropeni; < 1500/mm3, trombositopeni; < 100.000/mm3) gösterir. Pansitopeni olduğunda Kİ’inde tutulum oakla gelmekle birlikte LH’lerinde artış olmayabilir. Artmış hemafagositoz, bazen myelodisplazide pansitopeninin nedeni olarak eşlik edebilir.
2.2.4.7.Kulak Tutulumu
Hiçbir semptomu olmayan, ancak kulak akıntısı düzelmeyen her çocukta akla LHH gelmelidir. Sıklıkla temporal kemik tutulumuna eşlik eden bir bulgudur. İç, orta ve dış kulak ayrı ayrı veya beraberce tutulmuş olabilir. İçkulak tutulumu ani işitme kaybına neden olabilir. Sinüs ve nazal mukozada da tutulum olabilir. İçkulak tutulumu olmadıkça lezyonlara bağlı kalıcı sekel genellikle olmaz.
2.2.4.8.Endokrin Sistem Tutulumu
Sıklıkla diabet insipitus (arka hipofiz tutulum kaynaklı), boy kısalığı (%40 oranda; ön hipofiz tutulumu ya da kronik hastalığa- steroid kullanımına sekonder),hipotalamik infiltrasyona bağlı hiperprolaktinemi, hipogonadizm gibi endokrinopati bulguları görülebilir. (7) Nadir olarak pankreas ve tiroid tutulumu da bildirilmektedir.
2.2.4.9.Gastrointestinal Sistem Tutulumu
LHH’li olguların % 2-10’unda açıklanamamış ishal- kusma varlığında düşünülmelidir. Tanı GİS biyopsisi ile konur.
Tablo 2: LHH hastalığı tutulum sıklığı
Tutulan organ Tutulma yüzdesi Tutulan organ Tutulma yüzdesi
İskelet sistemi %80 Hematopoetik sistem % 15
Cilt %30-50 Akciğer %15
Hipofiz % 25 Lenf nodu %5-10
Karaciğer %15 SSS % 2-4
2.2.4.10.Santral Sinir Sistemi Tutulumu
SSS hastalığı, tutulan alanla ilişkili belirti ve bulgularla kendini gösterir ve buna bağlı olarak da geniş spektrumlu kinik ve laboratuar bulguları sergileyebilir. Şöyleki;
Hipotalamo-hipofizier sistem tutulumu olanlar; endokrinopati bulguları, Kafa içi yer kaplayan oluşma bağlı semptomu olanlar; baş ağrısı, KİBAS ve nöbetleri
Nörolojik disfonksiyonu- nörodejenerasyonu olanlar; refleks anormallikleri, ataksi, entelektüel disfonksiyon, tremor, hidrosefali bulguları,
Belirtilen semptomların birlikte görülebildiği olgular; kompleks bulguları, Radyolojik olarak saptanmış hastalık bulgusu olmasına rağmen, klinik bulgusu olmayan hastalık grubu şeklinde karşımıza çıkabilirler. (20, 28)
Bugüne kadar LHH’li olgularda SSS tutulumu ilişkili olarak tanımlanan belirti ve bulgular aşağıda görüldüğü gibi oldukça geniş spektrumdadır;
Etiyolojisi açıklanamayan baş ağrısı Görme problemleri
Agresivite
Hafıza problemleri Motor epileptik nöbetler Ataksi, dizartri
İntrakraniyal hipertansiyon Duygusal labilite
Sosyal davranış problemleri Obsesif kompulsif duygu durum Öğrenme güçlüğü
konsantrasyon kaybı
kekemelik, el yazı bozukluğu kendine zarar verme davranışı Hiperkinetik hatalık bulgusu Absans,
Epileptik nöbet Vertigo
Anormal refleks Nistagmus Hemiparezi Entelektüel kayıp Uyku bozukluğu Mide bulantısı Baş dönmesi Bulanık görme
Bunların yanı sıra bu olgularda klinik bulgu olmaksızın, SSS hastalığının radyolojik bulgularının saptanabildiği belirtilmektedir.(20)
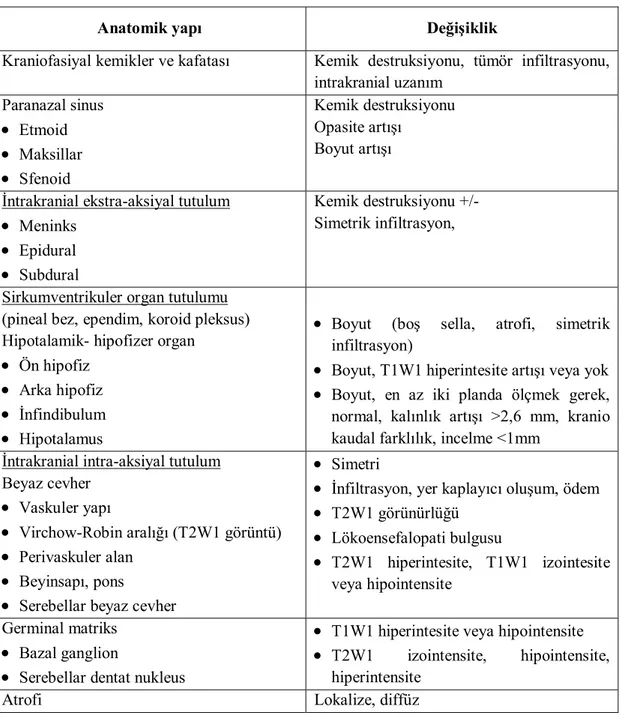
LHH’lu olgularda SSS hastalığı bulgularını tanımlayıcı araştırmaların son 2 dekatta artmakta olduğu görülmektedir. Bugüne kadar SSS bulguları olarak kesinleşmiş bir sınıflama tanımlanamamıştır. Giderek artış gösteren hasta sayıları ile yapılan çalışma sonuçları doğrultusunda tanımlama yöntemleri geliştirilmeye devam etmektedir. Geçmişten bugüne bakıldığında bu konuda önemli ölçüde olan hasta sayıları ile yapılan çalışmaların Grois ve arakadaşlarına ait olduğu görülmektedir. İlk veriler 1994 yılına ait 23 olgu ile tanımlanmış, 1998 yılında 38 olgunun MRI bulguları sınıflandırılmaya çalışılmıştır. Aynı çalışma grubu hasta sayısını arttırarak 2004 yılında 163 hasta ve 474 MRI görüntüsü ile hastalık bulgularını değerlendirmiştir. Elde edilen sonuçlarla SSS tutulumu patofizyolojisini açıklamaya çalışılmış ve lezyonların radyolojik olarak tanımlayıcı özellikleri oluşturulmaya çalışılmıştır. Bu çalışmanın sonucunda LHH, SSS tutulumu görüntüleme bulguları sınıflandırılabilir (Tablo 3).
Tablo 3: LHH, SSS tutulumu görüntüleme bulguları
Anatomik yapı Değişiklik
Kraniofasiyal kemikler ve kafatası Kemik destruksiyonu, tümör infiltrasyonu, intrakranial uzanım Paranazal sinus Etmoid Maksillar Sfenoid Kemik destruksiyonu Opasite artışı Boyut artışı İntrakranial ekstra-aksiyal tutulum
Meninks Epidural Subdural
Kemik destruksiyonu +/- Simetrik infiltrasyon,
Sirkumventrikuler organ tutulumu (pineal bez, ependim, koroid pleksus) Hipotalamik- hipofizer organ
Ön hipofiz Arka hipofiz İnfindibulum Hipotalamus
Boyut (boş sella, atrofi, simetrik infiltrasyon)
Boyut, T1W1 hiperintesite artışı veya yok Boyut, en az iki planda ölçmek gerek,
normal, kalınlık artışı >2,6 mm, kranio kaudal farklılık, incelme <1mm
İntrakranial intra-aksiyal tutulum Beyaz cevher
Vaskuler yapı
Virchow-Robin aralığı (T2W1 görüntü) Perivaskuler alan
Beyinsapı, pons
Serebellar beyaz cevher
Simetri
İnfiltrasyon, yer kaplayıcı oluşum, ödem T2W1 görünürlüğü Lökoensefalopati bulgusu T2W1 hiperintesite, T1W1 izointesite veya hipointensite Germinal matriks Bazal ganglion
Serebellar dentat nukleus
T1W1 hiperintesite veya hipointensite
T2W1 izointensite, hipointensite, hiperintensite
Atrofi Lokalize, diffüz
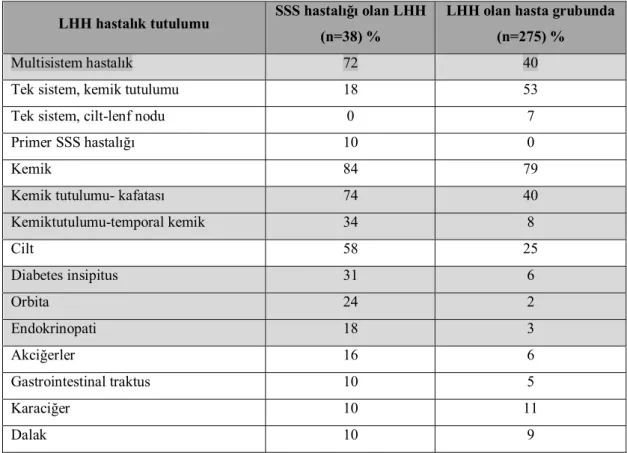
Prayer D, Grois N, Prosch H, Gadner H, Barkovich AJ. MR imaging presentation of intracranial disease associated with Langerhans cell histiocytosis SSS hastalığı olanlar sıklıkla, multisistemik hastalık ya da kafatası kemiği tutulumu olan olgulardan oluşmaktadır. 1998 yılında Grois ve ark.(21) ları LHH’lu olgularda multisistem hastalığın olması, kafatası kemikleri, temporal kemik tutulumu, orbita tutulumunun bulunması, diabetes insipitus ve endokrinopatinin saptanmasının SSS hastalığı gelişme riskini arttırdığını belirtmiştir. (21) LHH hastalık tutulan organ ve SSS hastalık birlikteliği Tablo 4’de verilmiştir. Bu alanların dağılımına bakıldığında kan beyin bariyerinin olmadığı alanlarda sıklık göstermesi dikkat çekicidir.
Yine aynı çalışmada, 38 SSS tutulumu olan hastaların kranial manyetik rezonans görüntüleme (MRG) bulgularına göre SSS lezyon sınıflaması yapılmış ve sıklık sırasına göre beyaz cevher tutulumu, gri cevher tutulumu, parsiyel boş sella, ekstraparankimal dura lezyonu, infundibular kalınlık artışı ve diffuz atrofinin saptandığını belirtmişlerdir (21).
Tablo 4: LHH hastalığı tutulan organ ve SSS hastalığı birlikteliği LHH hastalık tutulumu SSS hastalığı olan LHH
(n=38) %
LHH olan hasta grubunda (n=275) %
Multisistem hastalık 72 40
Tek sistem, kemik tutulumu 18 53
Tek sistem, cilt-lenf nodu 0 7
Primer SSS hastalığı 10 0
Kemik 84 79
Kemik tutulumu- kafatası 74 40
Kemiktutulumu-temporal kemik 34 8 Cilt 58 25 Diabetes insipitus 31 6 Orbita 24 2 Endokrinopati 18 3 Akciğerler 16 6 Gastrointestinal traktus 10 5 Karaciğer 10 11 Dalak 10 9
Grois NG, Favara BE, Mostbeck GH, Prayer D. Central nervous system disease in Langerhans cell histiocytosis. Hematol/Oncol Clin N Am 1998;12:287–305
Benzer diğer çalışmaların sonucunda SSS hastalığının MRG bulguları aşağıdaki gibi bildirilmiştir (21);
Kraniyofasiyal kemik lezyonları ve/veya yumuşak dolu uzantısı olan/olmayan kafatası tabanı tutulum bulguları
İntrakraniyal, ekstraaksiyal değişiklikler (hipotalamo-hipofizer değişiklikler, meninks, sirkumventrikuler organlar; epifiz, koroid pleksus)
İntrakraniyal-intraaksiyal değişiklikler (beyaz-gri madde tutulum, simetrik nörodejeneratif değişiklikler)
Literatürde LHH olgularında SSS hastalığının nöropatolojik bulguları, olgu bazında ilk 1956 da Freigin, 1956 da Kristensson,1966 da Yamaguchi tarafından tanımlanmıştır. Kapsamlı ilk çalışma 1979 yılında Kepes tarafından otopsi materyalinde yola çıkılarak sunulmuştur. (22)
Histopatolojik olarak SSS tutulum bulgularının tanımı yapılan çalışma sıklığı arttıkça değişmektedir. Son 3 dekatta artan hasta sayılarını içeren çalışmaların sonuçlarıyla tanımlar değişmekte ve gelişmektedir. SSS hastalığının saptandığı ilk yıllarda yapılan tanımlamaya göre SSS tutulum histopatolojisi 4 tipte incelenebilmektedir; (7)
o Hiperplastik proliferatif o Granulamatöz
o Xantomatöz o Fibrozis
Hiperplastik proliferatif evrede lezyonlar tanısal LHH hücreleri içerir. Serebrum ve serebellumda, beyaz cevherde demiyelinizasyon görülebilir ve histiositlerin yokluğu ile Purkinje hücrelerinin destrüksiyonu olabilir. Plazma hücre infiltratlarıyla yoğun gliozis belirlenebilir.Granulomlar beynin bağ dokusu içinde sınırlı olarak, meninks ya da koroid pleksusta tümöral lezyonlar oluşturabilir. Langerhans granulomları CD1a-reaktif hücreler ve belirgin CD8+pozitif T hücre infiltrasyonu ile periferal granulomlara benzer yapı gösterir. Granulomlar CD1a reaktif histiositlerle SSS parankimine kısmen infiltre olabilir. Bu durum nöron, akson kaybı ve gliozis ile karakterize şiddetli nörodejenerasyon ve T hücreye bağlı inflamasyon ile ilişkilidir. Serebellum ve beyin sapını etkileyen, CD1a hücre infiltrasyonu olmayan nörodejeneratif lezyonlarda CD8 reaktif lenfositlerle oluşan belirgin inflamasyon vardır ve doku dejenerasyonu, mikroglial aktivasyon ve gliozis ile ilişkilidir. LHH’de nörodejenerasyon T hücre ile oluşan inflamasyonla oluşur ve paraneoplastik ansefalite benzeyen sekonder demyelinizasyon, nöronal ve aksonal destrüksiyon ile karakterizedir.
2005 yılında Grois ve ark.(22) ları SSS bulgularından alınan 12 biyopsi örneğini inceleyerek histopatolojik olarak 3 farklı grup tanımlamışlardır;
1- SSS bağ doku içinde sınırlandırılmış granulomlar: geniş ekstra aksiyalalanda yerleşmiş, sıklıkla meninks ve koroid pleksusda yerleşmiş çoklu nodul olarak tanımlanmıştır. Bu noduler lezyonların, SSS dokusundan keskin sınırlarla ayrılmış olduğu, histiosit, köpüksü makrofaj, plazma hücresi, eosinofil, lenfosit
karışımı hücre gruplarından oluştuğu gösterilmiştir. Granulokm içi hücre oranları, LHH periferik organ granulomlarına benzer yapıda bulunmuş olmasına rağmen, SSS lezyonlarında saptanan T hücresi CD3+/CD8- yapısında olduğu saptanmıştır. CD1a + hücrelerinin dağılımı ve yerleşiminin farklılık gösterdiği, hatta bazı lezyonların belli kısımlarında saptanamadığı belirtilmiştir.(22)
2- SSS bağ doku içinde yerleşmiş ve infiltratif özellikte olan lezyonlar: Çalışmada iki olguda serebellar ve hipotamaik tumöral granulamatöz lezyon saptanmıştır. Bu grup granulomların özellikle CD1a+ histiosit, CD 68 makrofaj, T-B hücre ile infiltre olduğu görülmüştür. Lezyon yerine uyan alanlarda nöron ve aksonal kayıp olup, lezyon periferinde yerleşimli glial skar dokusu saptanmıştır.
3- Nörodejenetarif lezyonlar : bu grup lezyonlarda tüm beyni etkileyen dağınık yerleşimli, perivaskuler yerleşimli, CD8+ kaynaklı diffüz inflamasyon saptanmıştır. Bu alanlarda sitotoksik özellikte olan bu hücrelerin aktivasyonunda ek olarak, MHCI, MHCII eksprese eden, fagosit aktivasyon markırı taşıyan mikroglia aktivasyonu olduğu gösterilmiştir. Lezyon alanlarında granulom ve CD1a+ histiosit saptanmamıştır. Doku dejenerasyonunun yoğun olduğu alanlar serebellum, pons, mezensefalon, bazal ganglion, infindibulum olarak görülmüştür. Tanımlanan nörodejenerasyon ise en sık ve en aktif serebellumda saptanmıştır.
SSS hastalığının sık tutulum yeri olan hipotalamo-hipofizer sistem tutulumunda;
Hipotalamik tutulumla birlikte davranış değişiklikleri, iştah, vücut ısı, uyku bozukluğu
Posterior hipofiz tutulumunda, diabetes insipitus, poliüri, polidipsi
Ön hipofiz tutulumunda, büyüme gecikmesi, puberte prekoks- puberte tarda, amenore, hipotiroidi bulguları olabilmektedir.
Diabetes insipitus SSS hastalığının en yaygın klinik tutulumudur. Hastalığın derecesi ve aktivasyonu ile değişmekle birlikte %5-35 oranında sıklıkla görülmektedir. Dİ tanısı, susuzluk testi, MRİ da hipofiz sapı kalınlık artışı (> 2,5 mm), T1 ağırlıklı görüntülerde, arka hipofizde hiperintensite azalması ile saptanabilir. (7) İzole ön hipofiz tutulumu olan LHH lu olgular olabildiği gibi, bu olguların hipofiz tutulumu yapan SSS hastalıklarından (germinal tm vb gibi) ayrımın yapılması önemlidir.
Literatürde multisistem tutulumlu LHH hastalarında tanı anında DI oranının artma gösterdiği bildirilmektedir. 15 yıllık izlem sürecinde kafatası kemiği özellikle
göz, kulak, ağız tutulumu olanlarda Dİ gelişme olasılığının arttığı ve bu lezyonların SSS hastalığı için riskli tutulumu bölgeleri olduğu kabul edilmektedir (27) .
Bunun yanında LHH’li olgularda klinik olarak poliüri, polidipsi bulgusu olan, ancak susuzluk testi veya MRI bulgularıyla hipofizier patoloji saptanamayan olguların da var olduğu gösterilmiştir. Bu olguların da desmopresin sağaltımından yarar gördüğü gösterilmiştir (23). LHH sağaltım protokollerine bakıldığında, Dİ sağaltımında etkinliği yüksek modalite bulunmamaktadır. İzole olgular nadiren sistemik kemoterapiden yarar gören olgular olarak tanımlanmakta ancak bu kliniğin geri dönüşümsüz olduğu bilinmektedir. Kraniyal ışınlama yapılmış olan olgularda ise, radyoterapinin yan etkileri üzerinde daha çok durulmaktadır.
SSS hastalığı olarak ikinci sıklıkla etkilenen alan serebellar bölge olarak gösterilmektedir. Klinik bulgular da buna bağlı olarak, refleks değişiklikleri, ataksi, nistagmus..olarak görülmektedir.
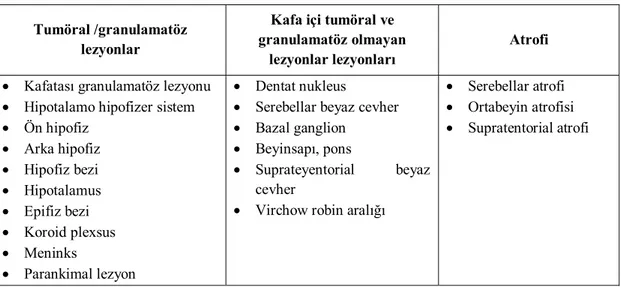
Histiocyte Society Group’un 2003 yılında LHH-SSS hastalığı ile ilgili tanısal bir rehber oluşturmuştur (24). Son 15 yılda elde edilen verilerini özetlenen 2010 yılında Grois ve ark.(23)’larının 308 SSS hastalığı olan hasta ve 935 kaniyal MRG’yi içeren çalışmasının sonucunda yapılan MRG’daki bulguların sınıflaması Tablo 5’de görülmektedir (23).
Bugüne kadar bildirilen tüm çalışmalarda amaçlanan, SSS hastalığı patofizyolojisini anlayabilmek, radyolojik olarak hastalık bulgularını ve görünümünü tanımlamak ve sonunda sık olarak progressif gidişli olan tutulumu durdurmaya yönelik sağaltım yaklaşımlarını belirlemeye yönelik olmaktadır.
Tablo 5: LHH- SSS hastalığında kraniyal MRG bulgularının sınıflaması (23). Tumöral /granulamatöz
lezyonlar
Kafa içi tumöral ve granulamatöz olmayan
lezyonlar lezyonları
Atrofi Kafatası granulamatöz lezyonu
Hipotalamo hipofizer sistem Ön hipofiz Arka hipofiz Hipofiz bezi Hipotalamus Epifiz bezi Koroid plexsus Meninks Parankimal lezyon Dentat nukleus
Serebellar beyaz cevher Bazal ganglion
Beyinsapı, pons
Suprateyentorial beyaz cevher
Virchow robin aralığı
Serebellar atrofi Ortabeyin atrofisi Supratentorial atrofi
2.2.5. LHH’da Tanı ve Tedavi Öncesi Klinik Değerlendirme
LHH’da sınıflama ve hastalık yaygınlığı, risk organ tutulumunun belirlenmesi gerekmektedir. Uygulanacak sağaltım tek sistem-tek odak dışında başlangıçta benzer olmakla beraber risk organlarına yönelik bazı ek sağaltım modalitelerinin belirlenmesi çok önemlidir.
LHH’un kesin tanısı klinik, laboratuar, radyolojik bulguların yardımıyla, lezyon dokunun histolojik ve immunofenotipik değerlendirmesi ile konmaktadır. Ancak bazı organ tutulumlarında, anatomik yerleşim yeri ile ilişkili biyopsi yapmak güç olabilmektedir. Bu durumlarda klinisyenin ayırıcı tanıda dikkatli olması gerekmektedir. Örneğin izole vertebra tutulumu, odontoid çıkıntı tutulumu gibi, hipofiz sapında kalınlaşma saptanan ve biyopsi yapılamayan olgular 3-6 ay sonraki izlemlerinde tekrar dikkatle değerlendirilmelidir.
Yukarda belirtilen bulguları olan olgularda, tanıda LHH düşünülmesi durumunda, tanıya yönelik laboratuar ve radyolojik değerlendirmede bazal yapılması gereken tetkikler:
Hemogram
o Hemoglobin, beyaz küre sayısı, periferik yayma bulgusu, trombosit sayısı Kan biyokimyası
o Total protein, albumin, bilurubin, ALT (SGPT), AST (SGOT), ALP, GGT o BUN, kreatinin, elektrolit
o Ferritin
Koagulasyon parametreleri o INR/ PT, APTT/PTT, fibrinojen Sabah ilk idrar bakısı
o Dansite, osmolalite Karın ultrasonu
o Karaciğer ve dalak boyutu ve parankim yapısı Akciğer radyografisi
Kemik survey radyografisi Kemik sintigrafisi
Elde edilen klinik bulgu ve laboratuar verileri sonucunda olası sistem tutulumunu saptamak üzere daha ayrıntılı inceleme yapmak gerekmektedir (Tablo 6). (29)
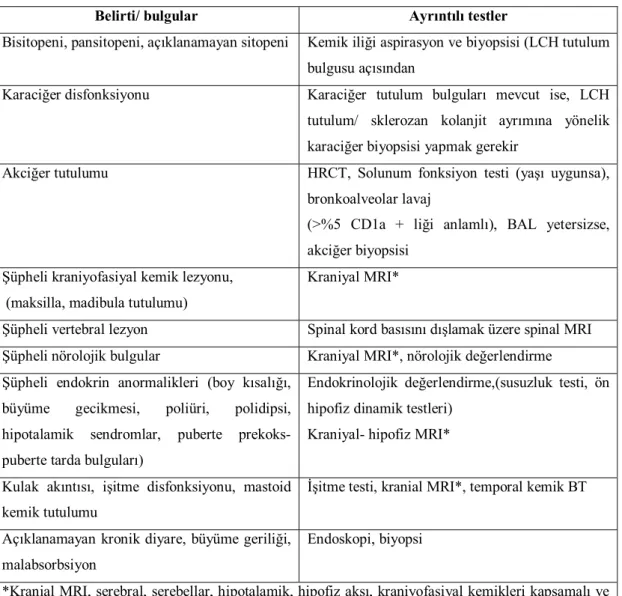
Tablo 6: LHH düşünülen olgularda sistem tutulumu saptamaya yönelik yapılması önerilen tetkikler Belirti/ bulgular Ayrıntılı testler
Bisitopeni, pansitopeni, açıklanamayan sitopeni Kemik iliği aspirasyon ve biyopsisi (LCH tutulum bulgusu açısından
Karaciğer disfonksiyonu Karaciğer tutulum bulguları mevcut ise, LCH tutulum/ sklerozan kolanjit ayrımına yönelik karaciğer biyopsisi yapmak gerekir
Akciğer tutulumu HRCT, Solunum fonksiyon testi (yaşı uygunsa),
bronkoalveolar lavaj
(>%5 CD1a + liği anlamlı), BAL yetersizse, akciğer biyopsisi
Şüpheli kraniyofasiyal kemik lezyonu, (maksilla, madibula tutulumu)
Kraniyal MRI*
Şüpheli vertebral lezyon Spinal kord basısını dışlamak üzere spinal MRI Şüpheli nörolojik bulgular Kraniyal MRI*, nörolojik değerlendirme Şüpheli endokrin anormalikleri (boy kısalığı,
büyüme gecikmesi, poliüri, polidipsi, hipotalamik sendromlar, puberte prekoks-puberte tarda bulguları)
Endokrinolojik değerlendirme,(susuzluk testi, ön hipofiz dinamik testleri)
Kraniyal- hipofiz MRI* Kulak akıntısı, işitme disfonksiyonu, mastoid
kemik tutulumu
İşitme testi, kranial MRI*, temporal kemik BT Açıklanamayan kronik diyare, büyüme geriliği,
malabsorbsiyon
Endoskopi, biyopsi
*Kranial MRI, serebral, serebellar, hipotalamik, hipofiz aksı, kraniyofasiyal kemikleri kapsamalı ve intravenöz kontrast kullanılmalı
LHH sağaltımı planlanırken, önemli iki noktayı vurgulamak gerekir:“ riskli organ tutulumu” ve “ SSS hastalığı açısından riskli tutulum yeri” saptanması tedavi planını, hastalık surveyi ve prognozunu, hasta izlem planının değiştirmektedir. Bu noktada “ riskli organ tutulumu” ve “ SSS hastalığı açısından riskli tutulum yeri” tanımları ve kapsamlarının açıklanması önemli olacaktır.
Riskli organ tutulumu
Hematopoetik sistem tutulumu:
o Hafif tutulum: (her iki bulgunun da olması gerekir) Anemi :hb 7-10 gr/dl
Trombositopeni : trombosit 20.000-100.000 mm3 o Ağır tutulumu: (her iki bulgunun da olması gerekir) Anemi :hb <7gr/dl
Trombositopeni : trombosit <20.000mm3 Karaciğer tutulumu:
Hepatomegali, >3 cm (kot altı, midklavikular hattan) Yetmezlik bulguları: o Hiperbilurubinemi o Hipoalbuminemi o GGT yüksekliği o Alt/Ast yüksekliği o Asit, ödem Dalak tutulumu:
o Splenomegali, >3 cm (kot altı, midklavikular hattan) Riskli olmayan organ tutulumları:
Akciğer tutulumu
Tek sistem tutulumu olup sistemik tedavi alması gereken grup İzole SSS riskli lezyonu olan
Multifokal kemik tutulumu olan grup SSS tutulumununun araştırılmasında;
İzole tutulum veya multisistem hastalıkla birlikteliği olabilir.
Kraniyal /spinal MRG (hipotalamo-hipofizer sistem, pineal bez, meninks, koroid plexus yanısıra parankimal dejenerasyon veya tumoral lezyonlar) değerlendirilmelidir. MRG’de ki özellikler aşağıda görülmektedir;
Kontrastlı, ince aksiyal T1 ağırlıklı kesitler
Hipotalamo-hipofizer alan için ince koronal ve sagital T1 ağırlıklı sekanslar (≤3 mm kalınlığında kesitler)
Tüm beyin için aksiyal T2 ağırlıklı, FLAİR sekansı (≤5 mm kalınlığında kesitler)
Beyin ve hipotalamo-hipofizer alan için kontrastlı, koronal ve sagital T1 ağırlıklı sekanslı çekim önerilmektedir.
Nörodejeneratif SSS-LHH bulgularının radyolojik olarak derecelendirilmesi: MRI bulgularına göre 3 farklı klinik derecesi bulunmaktadır.
Tablo 7: Nörodejeneratif SSS-LHH bulgularının radyolojik olarak derecelendirilmesi
Bulgular Bulguların tanımı
Hafif derece bulgular T1 ağırlıklı, bazal ganglion ve dentat nukleusta sinyal artışı, Serebellar beyaz cevherde, dentat nukleusla ilişki gösteren, lokal yerleşimli, T2 ağırlıklı sekansta sinyal artışı
Orta derece bulgular Beyinsapı, talamus, corpus kallosum yerleşimli anormal sinyal artışı
Ağır derece bulgular Subkortikal ve periventrikuler beyaz ve gri cevher değişklikleri
Bazal nörolojik değerlendirilme özellikle serebellar bulguların ön planda olması nedeniyle buna yönelik değerlendirmenin yapılması önerilmekte olup “ ICARS,EDSS,ABC-2 ölçekleri “ uygulanarak puan kaybı belirlenmelidir.
Bazal nöropsikolojik değerlendirme “ zeka testi, verbal ve motor kapasitenin, hafıza ve beceri yetileri” saptanmalıdır.
Endokrin değerlendirme “ idrar osmolaritesi, gerekli ise, susuzluk testi, ön hipofiz hormon paneli (TSH, T4, FSH, LH) IGF1 bakılması önerilmektedir.
Görme fonksiyonları (strabismus, görme alanı, stokom), beyin sapı uyarılmış potansiyelleri “ elektroensefalogram (EEG), görsel uyarılmış potansiyeller (VEP), beyin sapı akustik uyarılmış potansiyelleri (BAEP)” değerlendirmeleri önerilmelidir.
Nörodejeneratif SSS tutulumu olan hastaların değerlendirilmesinde BOS bakısı (hücre- protein içeriği) özellikle izole extraaksiyal kitlesel lezyonu olan olgular da yapılmalıdır.
2.2.6. LHH’da Klinik Sınıflama
Son rehberlere göre LHH, tek sistem tutulumu ve çoklu sistem tutulumu olan LHH olarak ikiye ayrılmaktadır (Tablo 8). (29)
Tablo 8: LHH’de klinik sınıflama
Tek sistem LHH (TS-LHH) Tek organ/ sistem tutulumu (tek / çoklu odak) Kemik unifokal (tek kemik) veya multifokal (>1
fazla kemik tutulumu) Deri
Lenf nodu (LHH lezyonu derene etmemiş olan) Akciğerler
Hipotalamo/hipofizer/ Santral sinir sistemi Diğer (tiroid, timus..)
Multisistem LHH (MS-LHH) 2 veya daha fazla organ/ sistem tutulumu (Riskli organ tutulumu olan/ olmayan)
2.2.7. LHH’da Sağaltım
LHH sağaltımı yıllar içinde, hastalık bulguları ve tutulum yerlerinin tanımlanmalarına paralel değişiklik göstermiştir. Histiocyte Society Topluluğunun yürütmüş olduğu, çok merkezli çalışmalarla tanımlanan farklı LHH tedavi protokolleri oluşturulmuştur.
LCH-1 protokolu: 1991-1995 yılları arasında değerlendirilmiştir bu protokol sonucunda vinblastin ve etoposid tekli tedavisinin,MS-LHH hastalarında eşit derece etkin olduğu gösterilmiştir. Kötü prognozla ilişkili olarak riskli organ tutulumunun olduğu saptanmıştır.
LCH-II protokolu: 1996-2001 yılları arasında uygulanmıştır.Bu çalışma sonucunda 2 yaş altı hastalarda, riskli organ tutulumu olmayan grupta sağaltıma yanıtın iyi olduğu ve yaşın izole risk faktörü olmadığı, hastalık ilişkili mortaliteden risk organ(RO) tutulumunun sorumlu belirtilmiştir.
LCH-III protokolu (2001-2008 yılları) sonucunda ise hastalar klinik risk sınıflamasına göre 3 sınıfta değerlendirilmiş olup; Grup1; MS-LHH,RO(+) hastaları, Grup2; MS-LHH, RO (-) hastaları ve Grup 3 ise multifokal kemik tutulumu olan olguları kapsayan, sistemik tedavi alması gerek hastalardan oluşmaktadır.
Son 20 yılda yapılan çalışmalar sonucunda görülen, MS-LHH, RO (-) hasta grubunda vinblastin+ prednisolon tedavisi etkinliği oldukça iyi olup, bu hasta grubu için standart tedavi protokolu olarak kabul edilebilir. Ancak idame sağaltımlarının uzun süreli olmasının relapsları azaltığı göterilmektedir.
2.2.7.1. Tek sistem, tek odak kemik tutulumu sağaltımı
Hastalığın büyük sıklıkla tutulum gösteren formudur. Çoğunlukla basit küretaj sağaltım için yeterli olabilmektedir. Ancak tutulum gösteren kemiğin yeri ile ilişkili (ağırlık taşıyan kemik olması, vertebra plana, spinal kord basısı, ağrı, fonksiyonel kayıp oluşturucu nitelikte olması gibi) olarak radyoterapi, veya lezyon içi steroid uygulaması yapılabilir.
Bu sağaltım seçeneklerinin uygulanamayacağı riskli kemik tutulumlarında sistemik sağaltım gerekebilir.
2.2.7.2.Tek sistem, çoklu odak kemik tutulumda sağaltım
Bu grupta sıklıkla sistemik kemoterapi verilir. (steroid, vinblastin kombinasyonu)
2.2.7.3. Tek sistem, cilt tutulumu sağaltımı
Genelde sadece cilt tutulumu, tüm LHH içinde %5 düzeyinde görülmektedir. Sıklıkla yenidoğan döneminde görülür. Spontan regresyon olmakla birlikte, multisistem LHH forma dönüşüm de görülebilir. Cilt tutulumu tedavisinde topikal steroidlerin yararlı olmadığı görülmüştür. Bu olgularda topikal nitrojen mustard preparatları kullanıldığı görülmektedir.(30) Cilt tutulumuna yönelik diğer kullanılabilen tedaviler, oral düşük doz metotreksat, steroid + vinblastin, azatioprin, PUVA olarak tanımlanmaktadır, ancak etkinlikleri tartışmalıdır.
2.2.7.6. İzole diabetes insipitus ve hipofiz tutulumda sağaltım
İzole stalk – hipofiz tutulumu sonucu gelişen Dİ geri dönüşümsüz olup, öncelikle semptomatik olarak DDAVP (Desmopressin) uygulaması başlatılır. Eşlik eden endokrinopati varlığında gerekli hormon replasmanları ayarlanır. Literatürde 2-CDA, etoposide, radyoterapi ile tedavi denenmiş vakalar da tanımlanmaktadır.(31,32,24)
2.2.7.7. Santral sinir sistemi -Serebral lezyonların Sağaltımı
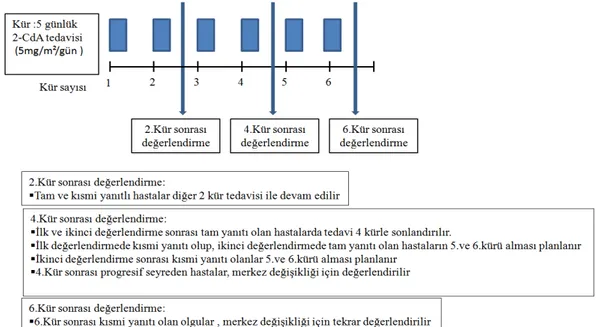
Hipofiz sapı lezyonlarına ek olarak, SSS lezyonlarının varlığında sistemik sağaltım endikasyonu bulunmaktadır. Kabul edilen standart sağaltımda steroid+ vinblastin (VBL) kombinasyonu, yanı sıra standart hatta yüksek doz immunglobulin uygulaması, 2-CdA, Cloforabin ± Ara-C ve radyoterapi olarak görülmektedir. (32,33). SSS-LHH hastalığı olan olguların sağaltımı aşağıda özetlenmiştir (şekil-2).
Şekil 2: SSS-LHH hastalığı olan olguların sağaltımı
Tablo 9: Tumöral SSS-LHH lu hastaların tedaviye radyolojik yanıt değerlendirilmesi
Yanıt değerlendirilmesi Yanıtın tanımı Tam yanıt SSS tumörün tamamen kaybolması
Kısmi yanıt Tek kitlede en uzun ölçümün en az %30 azalması Çoklu kitlede, toplam ölçümün en az %30 azalması Stabil hastalık Kısmi yanıtı ve ya progresif hastalık bulgusu olmayan
grup
Prograsif hastalık Tek kitlede en uzun ölçümün %20’den fazla artması Çoklu kitlede, toplam ölçümün %20’den fazla artması
Nörodejeneratif SSS-LHH hastalığı olan olguların tedavisi: izole radyolojik nörodejenrasyonu olan hastalarda klinik bulgu saptanmamış ise için tedavide görüş birliği henüz sağlanamamıştır.
o Sağaltım endikasyonu olan grup, “ klinik nörodejenerasyon bulgusu olup, radyolojik olarak da nörodejenerasyonun gösterildiği olgular” olarak tanımlamaktadır. Klinik bulgusu olmayıp, 6 ayda çekilen 3 MRda progresyon bulgusu olan hastaların, tedavisi hala tartışmalı olup buna yönelik araştırmalar devam etmektedir.
o Bu hasta grubunda 2 farklı modalitenin etkinliğinden bahsedilmektedir;
1-Ara-C: 12 ay devam edilen, aylık kürler halinde, kürde 5 günlük 150 mg/m2/gün dozunda Ara-C tedavi verilmektedir.
2-IVIG:12 ay devam edilen, aylık kürler halinde, 0,5 gr/kg/doz uygulanan IVIG verilmesi önerilmektedir.
Yanıt değerlendirmesi için;
1-MRG ile tedavi başladıktan 6.-12.24. aylarda kontrol, sonraki 5 yılda yıllık kontrollere devam edilmesi, hastalık stabil ise 2.5 yıl içinde iki yılda bir kontrolunun yapılması,
2-Nörolojik değerlendirmenin (ICARS,EDSS ile takip edilmesi) 6.-12 ve 24. aylarda kontrolu, sonraki 5 yılda yıllık kontrollere devam edilmesi, hastalık stabil ise 2.5 yıl içinde iki yılda bir kontrolunun yapılması önerilmektedir
3- Nöropsikiatrik değerlendirmenin 2 yılda bir tekrarlanması
4-Endorinolojik testler, idrar osmolaritesnin yıllık değerlendirilmeli,
5-Beyin sapı uyarulmış potansiyellerinin tedavi başladıktan 6.-12 ve 24. aylarda kontrolu, gerekli ise yıllık değerlendirilme,
6- BOS bakısının tedavi başladıktan 6.-12ve 24. aylarda kontrolu, gerekli ise yıllık değerlendirilme önerilmektedir.
Nörodejeneratif SSS-LHH da yanıt kriterlerinin değerlendirilmesi “İyi yanıt” kriterleri:
1-EDSS değerlendirmesinde, 5 adım kaybı 2-ICARS skorunda, 2 puan kaybı
3-MABC-2 ölçeğinde üst persentile geçme
4-Nöropsikiolojik değerlendirmede, klinik gelişme sağlanması 5-Radyolojik MRI bulguları
“kötü yanıt” kriterleri:
1-EDSS değerlendirmesinde, 5 adım artışı 2-ICARS skorunda, 2 puan artışı
3-MABC-2 ölçeğinde alt persentile düşme
4-Nöropsikiolojik değerlendirmede, klinik kötüleşme olduğu görülmesi 5-Radyolojik MRI bulguları
Tablo 10: Nörodejeneratif SSS-LHH’da radyolojik MRI bulgularıyla tedaviye yanıt değerlendirmesi
Yanıt katogorisi Regresyon Progresyon
Hafif Aynı durumda regresyon* Aynı durumda progresyon*
Orta
Regresyon gösteren durum değişikliği
(Ağır orta, orta hafif bulgulara geçiş gibi)
Progresyon gösteren durum değişikliği
(orta ağır, hafif orta bulgulara geçiş gibi)
3. GEREÇ ve YÖNTEM
Bu kesitsel ve geriye dönük olgu çalışması Ege Üniversitesi Tıp Fakültesi Çocuk Sağlığı ve Hastalıkları A.D. Çocuk Onkoloji Bölümünde Langerhans Hücreli Histiositoz tanısı ile izlenen olgularda SSS hastalığının sıklığını ve özelliklerini belirlemek üzere planlandı.
Çalışmanın hedefindeki 50 olguya ulaşıldı, ancak 30 olgu veya ailesi çalışmaya katılmayı kabul etti, 18 yaş altı olanların ailelerinden, 18 yaş üstünde olanların kendilerinden ayrıntılı onam alınarak çalışma başlatıldı.
Çalışmada SSS-LHH hastalığı bulgularını saptamaya yönelik hedeflenen, hastaların kranial MRG, EEG çekimlerinin yapılması, ayrıntılı nörolojik muayenelerinin ve gelişim/zeka gelişim testleri yapıldı.
Çalışma için Ege üniversitesi Tıp Fakültesi Etik kurulundan izin alınmıştır. (tarih 16.08.2012, No1396/596)
Hastaların Çocuk Onkoloji poliklinik izlem dosya kayıtlarından elde edildi. Tanı yaşı, tutulum yeri, tanı, izlem ve çalışma zamanındaki nörolojik bulgular, kranial MRG ları, çalışma zamanında EEG'leri, izlem bulguları kaydedildi. Çalışma kapsamında hastalar, Çocuk Nöroloji bölümünde nörolojik bakı ve EEG leri ve Radyoloji AD.'ında kranial MRG’ları elde edilerek ilgili öğretim üyelerince değerlendirildi.
Hastaların nörolojik gelişim basamakları, nörolojik hastalık lehine olabilecek belirtileri sorgulandı. Nörolojik muayenede, kranial sinir muayenesi, kas tonusu,kas gücü, refleks özellikleri, ayrıntılı serebellar sistem bulguları değerlendirildi. Davranış-dikkat-emosyonel durum değişiklikleri anamnezi alındı, okul başarısı sorgulandı.
Serebral epileptik odakları saptamak ve serebral bioelektrik aktiviteyi değerlendirmek amacıyla olgulara çocuk nöroloji EEG laboratuarında 40 dakika süre ile konvansiyonel EEG çekildi . Bipolar ve referans elektrot montajı yerleştirilmiş 10/20 uluslarası sistem kullanılarak, Nihon Kohden model EEG cihazı ile çekim yapıldı. Çekimler uyanıklık ve spontan veya sedasyon sağlanarak uykuda gerçekleştirildi. Provokasyon olarak hiperventilasyon ve fotik uyarı kullanıldı. Konvansiyonel EEG kayıtları çocuk nöroloji uzmanı tarafından, olgular hakkında klinik bilgi sahibi olmadan, kör olarak incelendi. Kayıtlarda hasta yaşına göre zemin ritmi ve interiktal aktiviteler değerlendirildi.
Onbir olguda gelişim ve zeka kapasitesini değerlendirmek amacı ile 6 yaş altı grubuna Ankara gelişim Envanteri, 6 yaş üstü olan 8 hastaya WISC-R zeka testi uygulandı. 1 hastanın psikiyatrik yakınmaları olması nedeniyle, o dönemde nörolojik ve psikiyatrik muayenesi yapıldı.
Ankara Gelişim Tarama Envanteri, 0-6 yaş arasındaki çocukların gelişimsel açıdan değerlendirilebilmesi amacıyla uygulanan bir envanterdir. AGTE, 0-6 yaş bebek ve çocukların şu andaki gelişimini ve becerilerini annelerden alınan bilgiler doğrultusunda değerlendirmektedir. Envanter, gelişimsel gecikme ve düzensizlik gösterme açısından risk altında olduğu düşünülen bebek ve çocukların erken dönemde tanınması ve gerekli önlemlerin alınabilmesine de olanak sağlar. Envanter “Evet, Hayır, Bilmiyorum” şeklinde yanıtlanan 154 maddeden oluşmuştur. Sorular gelişimin farklı, ancak birbiriyle ilişkili alanlarını (Dil-Bilişsel, İnce Motor, Kaba-Motor, Sosyal Beceri-Özbakım) temsil edebilecek biçimde düzenlenmiştir
WISC-R (Wechsler Çocuklar için Zeka Ölçeği- Revize Edilmiş Versiyonu) bireylerin zihinsel performanslarını belirlemek amacıyla uygulanan bireysel bir zeka testidir. Wisc-r testi sonucunda bireye ait sözel, performans ve genel olmak üzere üç zeka bölümü elde edilir. Wisc-r testinin bazı alt testleri süreye dayalı olarak uygulanır.
Gelişim testeri Tülay Aktaş Çocuk Onkoloji Bölüm psikologu tarafından yapıldı.
SSS hastalığı nöroradyolojik bulguları saptamak amacı ile hastaların kranial MRG'leri Radyoloji AD.'ında 3 Tesla MR G laboratuarında kontrastlı (IV gadopentetic acid içerikli madde) olarak Siemens Magnetoma Verio 3T (Erlangen/Almanya) cihazi ile çekildi. SSS bulguları saptamaya yönelik, ince kesitlerle, standart kraniyal MRG’a ek olarak hipofizer kesitleri de alındı.
Prekontrast görüntüler:
Aksiyal TSE T2 (TR/TE: 4000/186 msn, ST:5mm)
Aksiyal (Turbo inversion recovery) T1 (TR/TE/ TI : 3250/14/499 msn, ST:5mm)
Aksiyal TSE (500/10 msn, ST:5mm) Sagittal TSE T1 (450/10 msn, ST:5mm)
Coronal Dark Fluid (FLAIR) (TR/TE/TI 90000/92/2500, ST:5mm) Aksiyel SWI (27/20 msn ST:1,5 mm)
Aksiyel DWI B100(mm², sn) (5008/80, ST:5mm) Post kontrast görüntüler:
Aksiyal (Turbo inversion recovery) T1 (TR/TE/ TI : 3250/14/499 msn, ST:5mm)
Sagittal TSE T1 (450/10 msn, ST:5mm)
3D T1 A inversiyon recovery (MPRAGE) (TR/TE/TI 1590/ 2.24/900, ST:1mmm) Çekilen tüm kraniyal MRG’lar Nöroradyoloji öğretim üyeleri tarafından değerlendirildi. Kemik yapı ve yumuşak doku, dura, serebral parankim, bazal ganglion, serebellum, özellikle hipofizer yerleşimli olabilen patolojiler saptanmaya çalışıldı.
Santral nöropatolojik bulgular değerlendirilirken, Prayer ve ark(20), Duverneuil ve ark(35)’rının tanımlamış olduğu LHH kraniyal MRG bulguları göz önünde bulunduruldu.(20, 35,36,37,38,39,40,)
Literatürde SSS hastalık bulgusu olabildiği belirtilen genişlemiş venöz yapıların (dilate VRS) varlığı, pineal bez boyut artışı varsa not edildi. SSS herhangi bir alanda yerleşim gösterebilen kontrast tutan kitlesel lezyonlar, gir- beyaz cevherde T1/T2 sinyal değişikliği, nörodejeneratif, atrofi bulgularının varlığı sorgulandı. Özellikle hipofizer, bulgular açısından nörohipofiz sinyal yokluğu (bright spot yokluğu), hipofiz sap kalınlık artışı araştırıldı.
MRG görüntülerinde hipofizer bulgular için sap kalınlığı sagital ve koronal kesitlerde değerlendirildi. Maghnie ve ark(42)’larının tanımlamış olduğu boyutlara göre, Sap kalınlıkları ölçüldü. Hipofiz sap kalınlık ölçümlerine göre tanımlar:
Normal: <3mm,
Minimal kalın: 3-4,5 mm
Orta derecede kalın : 4,6-6,5 mm
Ağır kalınlık artışı: >6,5 mm (maghnie 18) olup İpliksi incelik: < 1mm olarak kabul edildi. (42)
Pineal bez değerlendirmesi yapılırken, sagital ve koronal kesitlerde maksimum çapı ölçülerek, Sumida ve ark(43)’larının tanımlarına uygun pineal bezin boyut artışı saptandı. Yaşa göre pineal bez boyutları normal aralığı
2. 2 yaş altında < 5,7 mm
Hastaların ulaşılabilen eski MRG ları’da tekrar değerlendirilerek, karşılaştırma yapılarak progresyon/ regresyon bulgusu değerlendirildi.
Bu çalışma Ege Üniversitesi Bilimsel Araştırma projesi (BAP) Komisyonu (proje no: 2013-TIP-90) tarafından ekonomik olarak desteklendi.
4. BULGULAR
1993-2014 yılları arasında izlenen toplam LHH tanısı alarak izlenen 67 olgunun ortalam tanı yaşı 74 ay (8- 192 ay) olup, ortanca yaş 72 ay dır. Olguların 39’i (%58,2) erkek, 28’i (%41,8) kız idi. Erkek/kız oranı 1,39’du. Olguların % 70’i tek sistem, %30’u multisistem tutulum göstermekteydi. İskelet sisteminin %94 oranı ile en sık tutulan sistem olduğu görüldü. Bunların %17 sinin diğer sistemlerle birlikte tutulduğu gözlendi. LCH- III protokolüne göre hastaların klinik sınıflamasında dağılım tek sistem/tek odak tutulumu olan %49 (33), tek sistem / çok odak tutulumu olan %21 (14), multisistem/ rikli organ tutulumu olmayan olguların %22 (15), multisistem/ rikli organ tutulumu olan olgular ise %8 (5) oranında saptandı.
SSS hastalığını araştıran 30 olgunun tanı yaş ortalaması 81,3 ay (8-192ay) olup ortanca 74 ay idi, 16’sı erkek, 14’ü kız olguydu. Küçük yaş olarak değerlendirdiğimiz 36 ay altı hasta sayısı, 11 olup %36’sı (11) 3 yaş altındaydı .Bu 11 hastanın, %63’ü (7) multisistem hastalığı olan olgulardı.
Tablo 13’de olguların tüm özellikleri görülmektedir. Bu grupta iskelet sisteminin büyük oranda tutulduğu görüldü (Tablo 11).
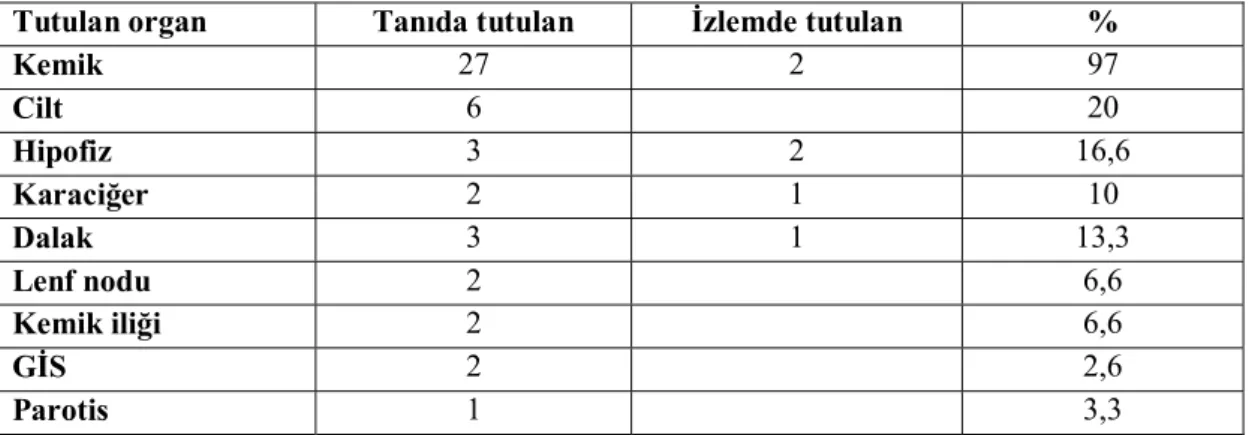
Tablo 11: SSS hastalığı araştırılan olgularda tutulan organların sıklığı
Tutulan organ Tanıda tutulan İzlemde tutulan %
Kemik 27 2 97 Cilt 6 20 Hipofiz 3 2 16,6 Karaciğer 2 1 10 Dalak 3 1 13,3 Lenf nodu 2 6,6 Kemik iliği 2 6,6 GİS 2 2,6 Parotis 1 3,3
LCH III’e göre 30 olgunun klinik sınıflandırılması tablo 12’da özetlendi.
Tablo 12: 30 olgunun klinik sınıflandırılması
Hastalık yaygınlığı n %
Tek sistem /tek odak/ 14 46
Tek sistem / çok odak 6 20
Multisistem /risk organ tutulumu olmayan (MSS-RO -) 6 20
Olgulardan birinde nörofibramatozis tip 1, LHH kliniğine eşlik ediyordu
LHH- SSS için riskli kemik tutulumu 7 hastada olup, bunların 6’sında temporal kemik tutulumu, 1’inde orbita tutulumu bulunmaktaydı.
Klinik bulgu olarak tanıda, 3 hastada Dİ kliniği ve bir hastada kazanılmış motor fonksiyonlarında gerileme (yürüme ve konuşma gerilik) saptanırken, izlemde 2 hastada Dİ geliştiği ve tanıda Dİ’li bir hastada da ek olarak nörodejeneratif ve psikiyatrik patolojiler olduğu gözlendi.
LCH III/IV protokolune göre sınıflandırılan olguların, 18’ine sistemik kemoterapi (prednisolone+vinblastin) uygulandı. 10 olguya küretaj ile lokal sağaltım yapıldı, 2 olguya radyoterapi verilmiş ve tüm olgular izleme alındı.
İzlemde 13 hastada relaps gelişti ve ortalama relaps zamanın 14 ay (3-28 ay) idi. İki olguda yineleyen relapslar olup, bunların bir tanesinde klinik ve radyolojik SSS hastalığı gözlendi.
Relaps gelişen olgularda radyoterapi, ikincil sağaltım ve kurtarma sağaltımı verildi ve 4 olguda akraba dışı allojenik hematopoetik kök hücre nakli (AHKHN) uygulandı.
Olguların nöropsikiyatrik değerlendirmelerinde, 1 olguda belirgin nörodejeneratif bulgulara ek psikiyatrik bozukluk saptanırken, 3 olguda da gelişim ve zeka testi bakılarında ılımlı gerilik vardı. Bu olgularda radyolojik ve klinik nöropatolojik bulgularla birliktelik gözlendi. (tablo14-15)
Tüm olguların EEG’lerinde patolojik bulgu saptanmadı.
Gelişim ve zeka testi toplam 11 hastaya uygulanabildi. 6 yaşın altıda 3 hastaya AGTE uygulanarak, 2 sinde gelişim gecikmesi saptandı. Bu hastalar 2 yaş altı multisistem tutulumlu olgulardı. WISC-R uygulanan 8 olgunun, 2’sinde sınırda zeka kapasitesi gözlendi. Bu olgulardan 1’i Dİ’li idi.
Kraniyal MRG’i incelenen edilen 30 olgunun % 33,3 (10)’unda, SSS hastalığı uyumlu bulgular gözlendi (Tablo 16) Bu olguların olguların klinik ve radyolojik bulguları tablo 13-14- ve 15’te görülmektedir . Bu MRG bulguları sırası ile
Nörohipofiz patolojisi (5 olguda) Stalk kalınlaşması (4 olguda) Genişlemiş VR aralığı (2 olguda) Serebral atrofi (1 olguda)
Serebellar ve beyin sapı atrofisi (1 olguda)
Pons ve mezensefalonda kontrast tutan lezyon (1 olguda)
Bilateral serebral hemisferde hipomyelinizasyon ve gliozis (1 olguda) Derin beyaz cevherde hiperintens lezyon ve izlemde gliozis (1 olguda) Pineal bezde patoloji (2 olguda) görüldü.
Bazı olgularda birden fazla patolojik MRG bulgusu bulunmaktaydı. Ayrıca hiçbir klinik bulgusu olmayan 7 olguda, kuşkulu MRG bulguları dikkat çekti.
Klinik ve radyolojik bulgu birlikteliği gösteren, 6 olgu “kanıtlı SSS hastalığı” olarak değerlendirildi(Tablo 16). Diğer 4 olgu, radyolojik görüntülemede patoloji yanı sıra, SSS hastalığı riski açısından organ tutulumu pozitif bulgular ve bazılarında nöropsikiyatrik bozukluk bulunması üzerine bu hastalar “ kuvvetle olası SSS hastalısı” olarak değerlendirildi(Tablo 16). Diğer olgu grubunda (7 olgu, C grubu) SSS hastalığı açısından çoğu risk taşımayan, hepsi tek sistem tutulumlu olan, 3 yaşından büyük, ancak kraniyal MRG’larında LHH lehine görüntüleri saptanan olgular olup “ SSS açısından kuşkulu” olarak düşünüldü (Tablo 16).