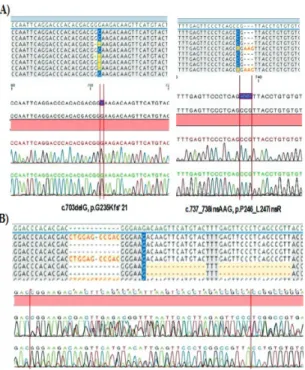
PTEN and AKT1 variations in childhood T-Cell acute lymphoblastic leukemia
Tam metin
Şekil
Benzer Belgeler
2013 年國際口腔雷射應用醫學會(SOLA)世界年會假北醫大盛大舉行,來自歐 美亞等國近 200 名專業人士與會
The use of mother tongue (L1) has been an inevitable part of second or foreign language teaching in various contexts where both the teachers and the learners have the same
[r]
BÜYÜK ÂFET, asıl adı Yoıjgiki olan b ü Hıtrvat delikanlısıydı; o zamanlar İstadbuMa Yorjgakisiz düğün olmaz gibiydi; tdk kusuru,R. •
Bu makalede, ilk olarak Râzî’nin bir Şâfiî âlim olarak portresi sunulacak, kendisinin mezhep âlimlerince ve tabakât yazarlarınca nasıl telakki edildiği, üzerine
aGVHD: Acute graft-versus-host disease; allo-SCT: Allogeneic hematopoietic stem cell transplantation; AML: Acute myeloid leukemia; ANC: Absolute nuclear cells; ATG:
The patient underwent surgical extraction of the large aortic vegetation, and a solitary mass measuring approximately 15x20 mm was observed in the subaortic
Ayaş tüneli inşaatı ihalesinde ya- pılan bütün hatalar bu kısımdaki işin ihalesin- de de tekrarlanmıştır, Arifiye - Sincan yeni demiryolu güzergahı üzerinde tahminen 60
