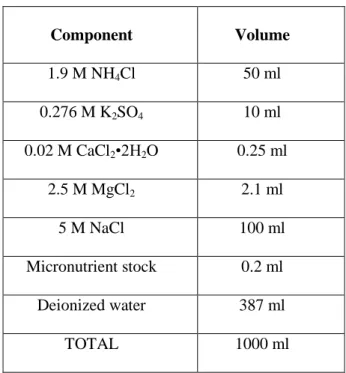
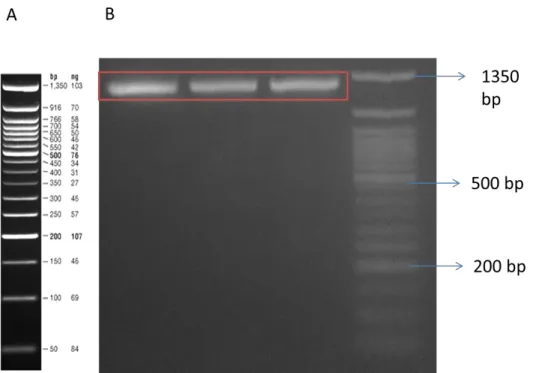
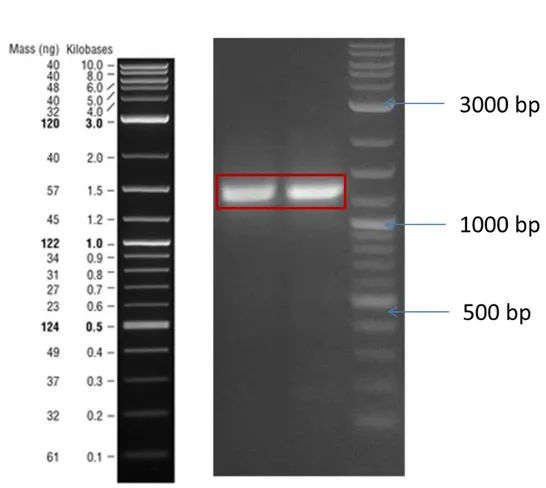
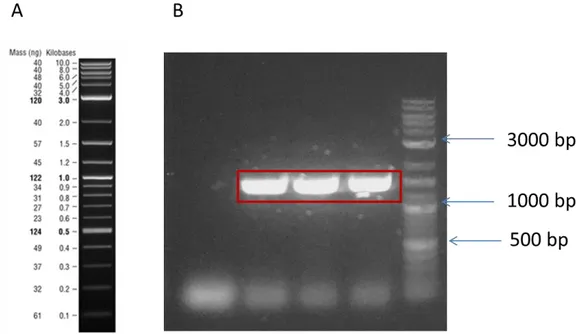
Developing synthetic biology enabled whole cell biosensors
Tam metin
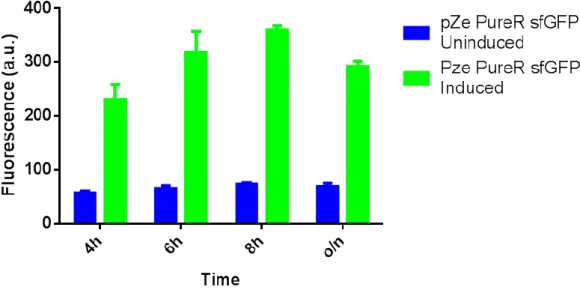
Şekil

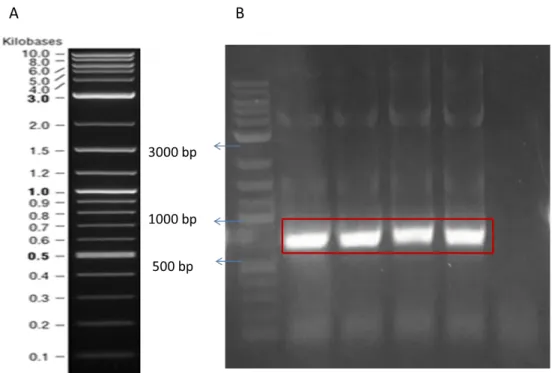
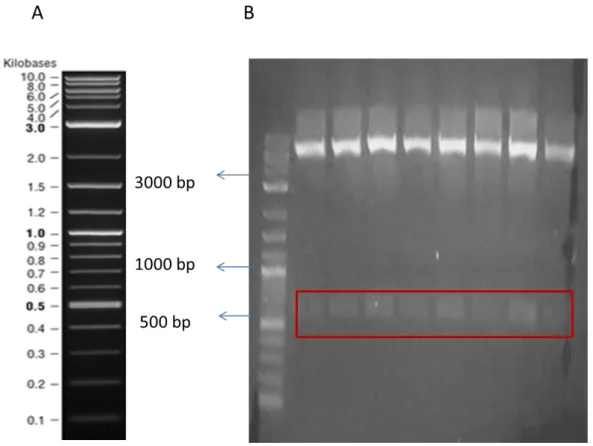
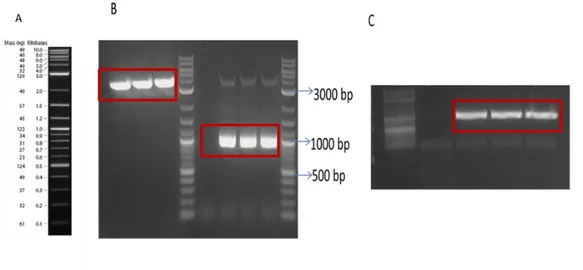
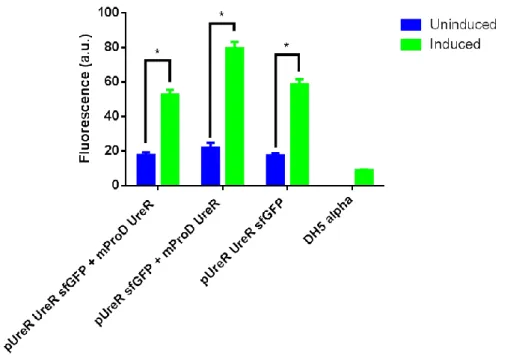
Benzer Belgeler
these systems become unstable when arbitrary small time delays were introduced into the feedback laws; see, e.g., [5] and [6]. This lack of robustness and some other
Heterogeneity within tumors is not only due to cellular plasticity but also their ability to alter tumor microenvironment through complex interaction of various
Yani “Bahçıvan bir gül için bin dikene katlanır.” Yaşamım okuyunca, Sakallı Celal’in bin dikenle yetinmediğini, katlanılacak başka dikenler peşine düştüğünü
To test the utility of the whole-cell model in this context, we experimentally measured the growth rates of 12 single-gene disruption strains— ten of which were correctly predicted
Figure 12-38 Molecular Biology of the Cell (© Garland Science 2008). Sinyal dizileri Pürüzlü
Yerleşim gerçekleştikten sonra Protein disülfit izomeraz tarafından serbest sülfidril gruplarının yükseltgenmesi sağlanır.. Sitozol tarafı ise indirgeyici koşullar
antikor çeşitliliğini sağlamakta kullanılan sistemler aynıdır, aynı V(D)J rekombinaz kullanılır, ancak somatik hipermutasyon görülmez. uyarılmaları Antijen sunan
By monitoring the bacteria contents of fish organs, the quality of fish can be measured since these will affect the storage life and quality of the fishery products (Kaneko,
