Molecular Mechanisms of Early and Late LTP
Tam metin
Şekil
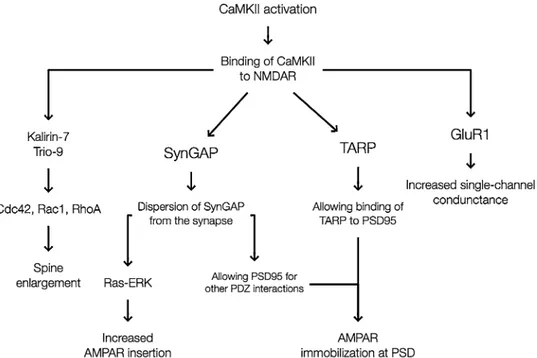
Benzer Belgeler
Similarly, correlation of mechanical noise can be calculated by using the generalized Nyquist theorem which states that the current noise correlation between two ports in an
The groups which are isomorphic to the auto- morphism group of such a compact bordered Klein surface with this maximal number of automorphisms are called M ∗ -groups.. Also, the
Düşünme, merak, sorma, ve araştırma kültürel edinimlerdir Uçan'ın da belirttiği gibi sanat olgusuna duyarlı bir toplum için ön koşullardır (Uçan, 1996). Düz
[r]
In contrast to arsenite treatment, activation of ERK1/2 was not detected in curcumin-treated colorectal carcinoma cells, andNAC and PD98059 did not show any inhibitory effect on
İstan bul Devlet Balesi dansçıları üç yıl boyunca yalnız operaların bale bölümlerinde rol alabildiler; bu nun nedeni İstanbul Devlet Öpe- ra ve Balesi'nin
the composition and diameter of fiber types in animal masticatory muscles. 1,3-5 In the present study, physiological stimulation through masticatory functional load revealed
Ülkemizde aile hekimliği disiplininin Dergimiz- le başlayan bilimsel yayın etkinlikleri 2016 yı- lının başlarında çok önemli noktalara gelmiş du-
