Cloning and characterization of α-amylase from Bacillus circulans ATCC 61
Tam metin
Şekil


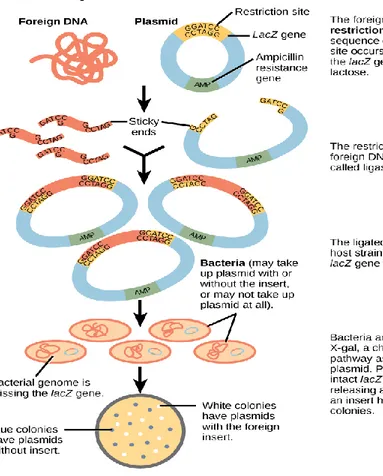


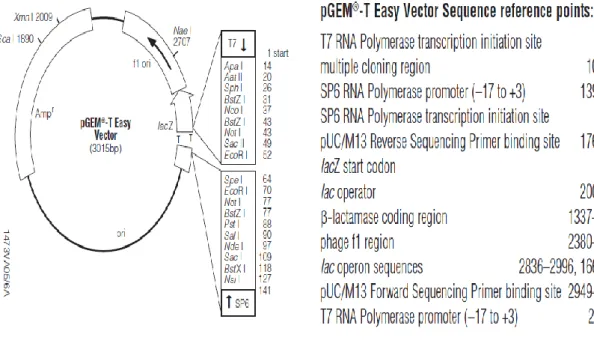

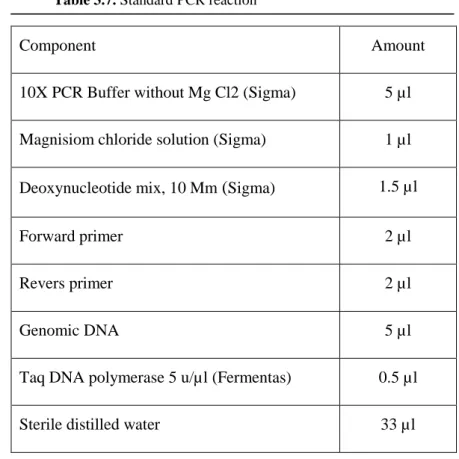

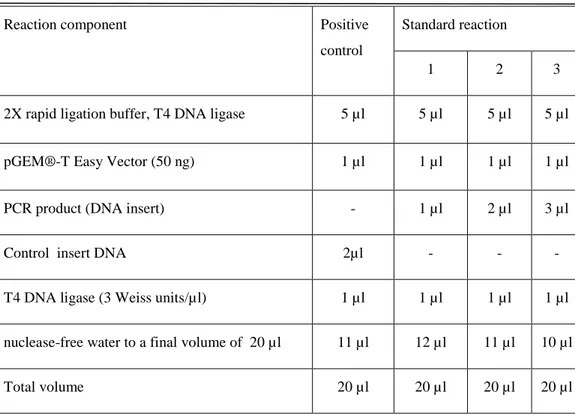
Benzer Belgeler
New treatment modalities such as new drugs or stem cell transplantation are required for children with risk organ positive multisystem disease, as the prognosis remains
Daha sonraları CFC’lerle birlikte bu soğutkanlara alternatif olarak üretilen HCFC ve HFC türü halokarbon soğut- kanların da küresel ısınmaya neden oldukları ortaya
sınıf öğrencilerinde öğretim yöntemi ve cinsiyetin, fen başarısı, mantıksal düşünme yeteneği ve yaratıcı düşünme yeteneği üzerinde ki etkilerini
Based on our previous experience, we used synthesized CMC/Chitosan- α-Fe 2 O 3 NPs due to their characteristic properties such as their nanoscale size, shape, high surface
For a clustering scheme employing a regular grid, where the box size is fixed for each level, symmetry of the translations leads to a significant reduction in the number of
In binary mode the source node starts with L copies of the message; any node A (source or relay) that carries n > 1 message copies, and meets another node B which does not have
Further, sequence and structure analyses showed a high structure and sequence similarity between dMT hinge region and the DNA binding domain of a cyanobacterial
The deduced amino acid sequences of mt-a and mt-d genes show striking similarity to the MT-like proteins described within the Class II as Type 1 MTs and showed 100 % similarity
