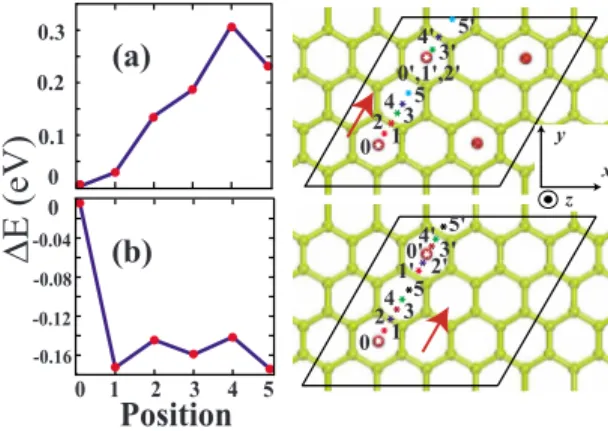
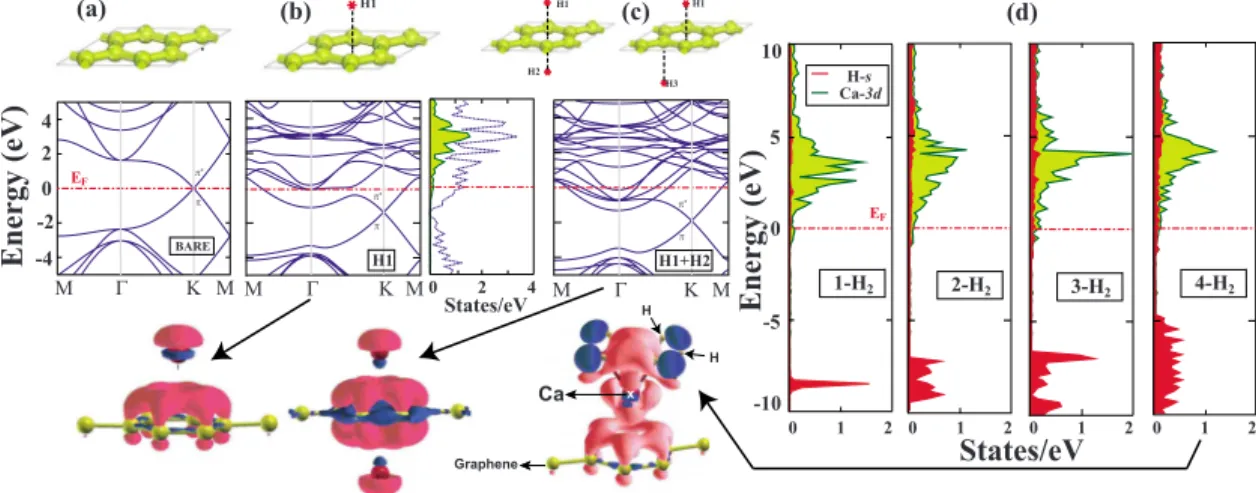
Hydrogen storage of calcium atoms adsorbed on graphene: First-principles plane wave calculations
Tam metin
Şekil

Benzer Belgeler
normalized rms.. Transient analysis is performed with SPICE and BUSTLE with a square wave input. The voltage waveform on the load can be seen in Fig. It is surprising
conventional relation between selling and buying prices is such that, the buying price is costly than the selling price to the carbon market. Let Q represent the production quantity
A self-financing trading strategy is called admissible if its value process is non-negative at the time of expiration of the contingent claim Fo¨llmer and Schied 2004, definition
Bu bildiride, veri matrisin boyutlarının yüksek oldu˘gu do˘grusal denklem sistemlerini çözmek için kullanılan, birinci dereceden yinelemeli çözülerin aksine, yakınsama
If we know (i) the execution plan of each query, (ii) an estimated cost for each operator/plan, and (iii) the target multicore platform in advance, we can suggest compile-
This kind of conducting polymer synthesis not only improves the poor mechanical and physical properties of heterocyclic polymers but also retains the con- ductivity to a
Abstract – We study theoretically the electronic transport in parallel few-level quantum dots in the presence of both intradot and interdot long-range Coulomb interaction.. Each dot
In this study, a design model was developed for the space planning of child care centers. In order to reach this aim, the child care centers were analyzed considering the
