i )ofaseriesofexperimentallytestedcompounds K Dockingstudiesonmonoamineoxidase-Binhibitors:Estimationofinhibitionconstants(
Tam metin
Şekil
![Table 1. AutoDock estimated free energies of binding (DG b ), calculated [K i (calculated)] and experimental [K i (experimental)] inhibition constants of the studied inhibitors (temperature = 298.15 K)](https://thumb-eu.123doks.com/thumbv2/9libnet/4322278.70839/2.892.67.818.899.1113/estimated-calculated-calculated-experimental-experimental-inhibition-inhibitors-temperature.webp)
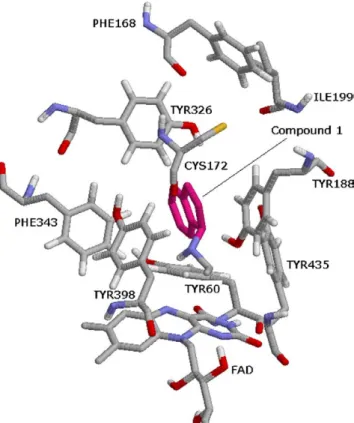
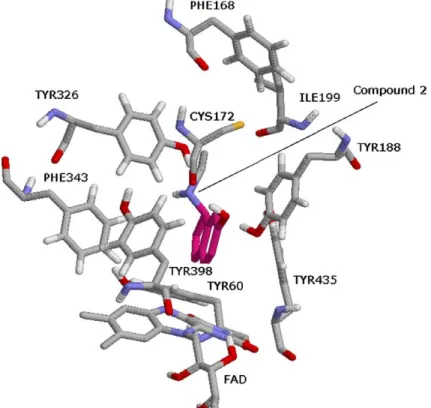
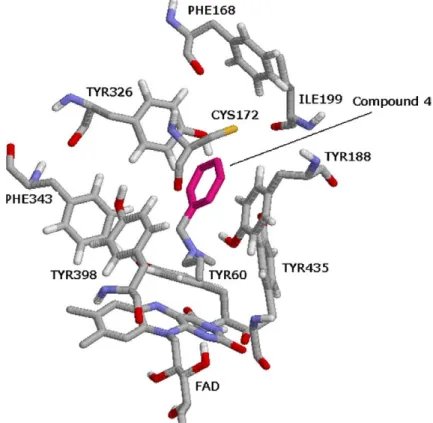
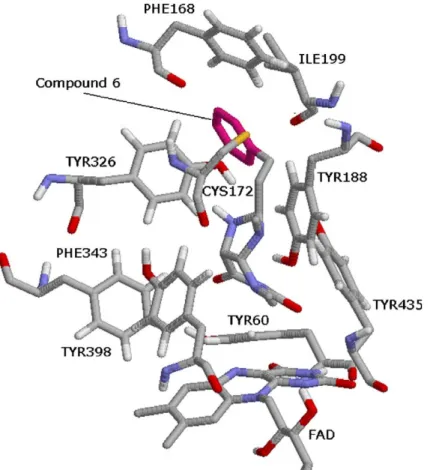
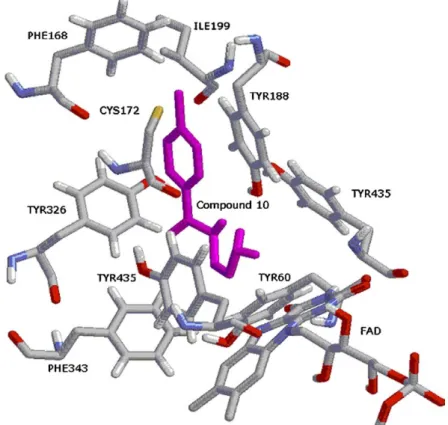
![Figure 9. The binding conformation of 3-methyl-8-(4,4,4-trifluoro- 3-methyl-8-(4,4,4-trifluoro-butoxy)indeno[1,2-c]pyridazin-5-one 9 with MAO-B (1S3E: 1.6 A˚ resolution).](https://thumb-eu.123doks.com/thumbv2/9libnet/4322278.70839/7.892.239.669.100.570/figure-binding-conformation-trifluoro-trifluoro-indeno-pyridazin-resolution.webp)
Benzer Belgeler
Su kaynakları, kooperatifler ve balık çiftliklerine ait ayrıntılı uzaysal veri tabanının oluşturulması ve CBS içinde değerlendirilmesi karar vericiler için
「99學年度新生入學指導」圓滿成功!
[r]
Nâzım Hikmeti iyi tanımak, iyi bilmek kendisine Türk aydını, Türk yurttaşı diyen herkesin görevidir demek istiyorum.. ‘İyi ta nımak’, sağlam,
Değişik yemekten hoşlananla- ra, yaratıcılığı sevenlere, düş kı rıklığına uğramamaları için “ Fırında Piliç” tavsiye ederim; piliç, lokantanın
Süreçte, öncelikle alt kriterlere göre oluşturulan karşılaştırma matrislerinin VZAHP ağırlıkları hesaplanmış ve Tablo 3’te maliyet ana kriterinin alt kriterlerine
The aim of this experiment is to find the operating voltage range of an Anthracene crystal detector and determine the optimal operating voltage for the further
Şekil 3 nokta yarıçapının fonksiyonu olarak sonlu sınırlandırıcı potansiyele sahip küresel kuantum nokta yapının taban ve çeşitli uyarılmış durumlarının
