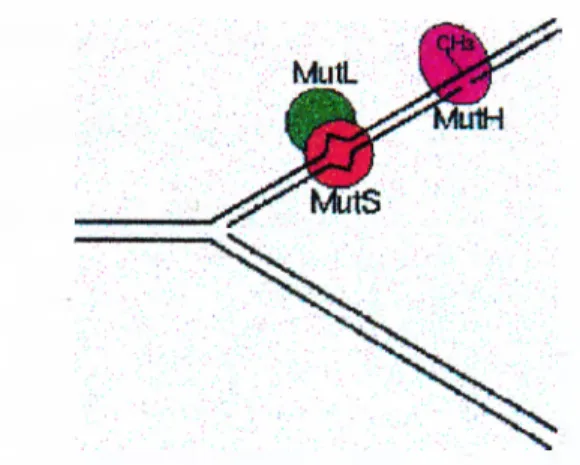
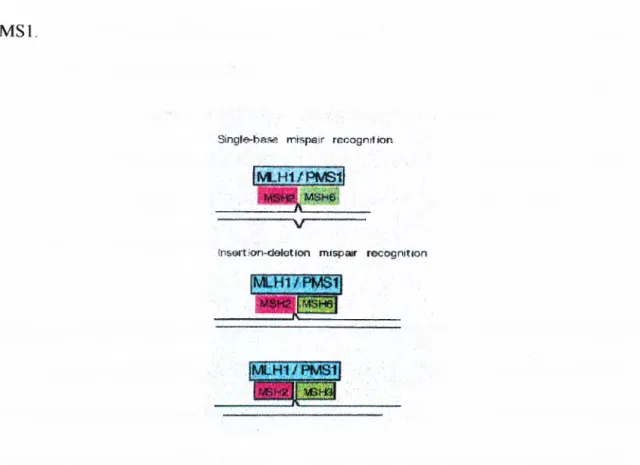
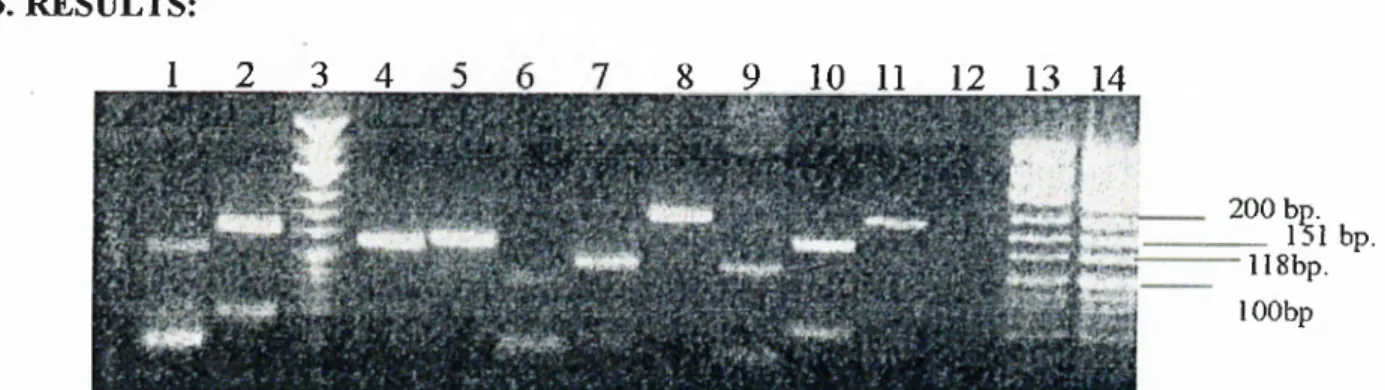
Development of a non-radioactive diagnostic test for the detection of microsatellite instability in colorectal cancer
Tam metin
Şekil



Benzer Belgeler
İlkin Abidin Dino'nun önayak ol masıyla 'Liman Sergisi'nde, daha son ra, hepsi de pırlanta bir resim kuşağını oluşturan 'Yeniler Grubu'nda dikkati
Олардың сол сенімді орындауы үшін оларға үлкен жол көрсетуіміз керек, сондықтан біз химия пәнін агылшын тілінде байланыстыра оқыту арқылы
Findings suggest that the proposed method has a promising and reliable tool for prostate cancer screening in clinical applications cost-effectively to help physicians for
One of the SSR markers (Xgwm382) located on chromosome group 2 (A, B, D genomes) was present in the resistant parent and the resistant bulk but not in the susceptible parent and
Kırsal alanlar nüfus yoğunluğunun az olduğu, ekonominin genel olarak tarıma dayalı ve sosyal imkânların yeterince gelişmemiş olduğu homojen alanlardır. Kırsal
Technological advances with the integration of imaging and com- puter software in every phase of radiotherapy include simulation, MLC modulation of radiation beams, improved
The pur- pose of the present study was to provide a prospective comparison and determine the validity of urine (leukocyte esterase, nitrites, microscopy, and urine culture) and
Conclusion: The use of a fecal occult blood test in colorectal cancer screening followed by a colonoscopy when there are positive test results is an effective and essential method
