T. C.
DİCLE ÜNİVERSİTESİ TIP FAKÜLTESİ
İÇ HASTALIKLARI ANABİLİM DALI
AKUT MİYELOİD LÖSEMİLİ HASTALARIMIZDA
t(15;17), t(8;21), inv(16), t(9;22)
TRANSLOKASYONLARININ SIKLIĞI VE PROGNOZA ETKİSİ
(UZMANLIK TEZİ)
Danışman: Prof. Dr. Orhan AYYILDIZ
Dr. Ergün PARMAKSIZ
T. C.
DİCLE ÜNİVERSİTESİ TIP FAKÜLTESİ
İÇ HASTALIKLARI ANABİLİM DALI
AKUT MİYELOİD LÖSEMİLİ HASTALARIMIZDA
t(15;17), t(8;21), inv(16), t(9;22)
TRANSLOKASYONLARININ SIKLIĞI VE PROGNOZA ETKİSİ
(UZMANLIK TEZİ)
Danışman: Prof. Dr. Orhan AYYILDIZ
Dr. Ergün PARMAKSIZ
TEŞEKKÜR
İç hastalıkları uzmanlığı eğitimim süresince yetişmemde büyük emekleri olan saygıdeğer hocalarım; Rektörümüz Sayın Prof. Dr. Fikri Canoruç’a, İç Hastalıkları Ana Bilim Dalı Başkanımız Sayın Prof. Dr. Ekrem Müftüoğlu’na, tez danışmanım Sayın Prof. Dr. Orhan Ayyıldız’a, Sayın Prof. Dr. Vedat Göral’a, Sayın Prof. Dr. M.Emin Yılmaz’a, Sayın Prof. Dr. Mithat Bahçeci’ye, Sayın Doç. Dr. Abdurrahman Işıkdoğan’a, Sayın Doç. Dr. Mehmet Dursun’a, Sayın Doç. Dr. Kendal Yalçın’a, Sayın Doç. Dr. Alparslan Tuzcu’ya, Sayın Doç. Dr. Şerif Yılmaz’a, Sayın Doç. Dr. Alı Kemal Kadiroğlu’na, Sayın Yrd. Doç. Dr. Dede Şit’e, Sayın Yrd. Doç. Dr. Abdullah Altıntaş’a, Sayın Yrd. Doç. Dr. Ramazan Danış’a, Sayın Yrd. Doç. Dr. Şenay Arıkan’a, Sayın Yrd. Doç. Dr. Kadim Bayan’a, Sayın Yrd. Doç. Dr. Şehmus Özmen’e, Sayın Yrd. Doç. Dr. Davut Akın’a, Sayın Yrd. Doç. Dr. Yekta Tüzün’e, Sayın Yrd. Doç. Dr. Deniz Gökalp’a, Sayın Yrd. Doç. Dr. Timuçin Çil’e, emekli öğretim üyelerimiz Sayın Prof. Dr. Bünyamin Işıkoğlu’na, Sayın Prof. Dr. Halil Değertekin’e, Sayın Prof. Dr. Orhan Yazanel’e, teşekkür ederim.
Rotasyon eğitimim sırasında bilgilerinden yararlandığım Kardiyoloji Anabilim Dalı öğretim üyelerine, Göğüs Hastalıkları ve Tüberküloz Anabilim Dalı öğretim üyelerine, Enfeksiyon Hastalıkları ve klinik Mikrobiyoloji Anabilim Dalı öğretim üyelerine ve Biyokimya Anabilim Dalı öğretim üyelerine teşekkür ederim.
Tezimin hazırlanmasına katkıda bulunan Hematoloji laboratuarından Kim. Murat Yurt’ta, Bio. Cahit İlhan’a, Sağlık Teknikerleri Emine Batum ve Mahmut Esen’e teşekkür ederim.
Birlikte çalıştığım tüm asistan arkadaşlarıma, İç Hastalıkları A.D. çalışanlarına ve gerek eğitimimin gerekse tezimin hazırlanışının her aşamasında desteğini esirgemeyen Sayın Doç. Dr. Alparslan Tuzcu, değerli arkadaşım Dr. Süleyman Sayar’a ve Uzm. Dr. Semir Paşa’ya ve her zaman yanımda olduklarını bildiğim aileme teşekkür ederim.
İÇİNDEKİLER Sayfa TEŞEKKÜR………. i İÇİNDEKİLER……… ii KISALTMALAR………. iii ŞEKİLLER DİZİNİ………. iv TABLOLAR DİZİNİ……… iv ÖZET……… v SUMMARY………... vi 1- GİRİŞ VE AMAÇ……… 1 2- GENEL BİLGİLER………. 3 3- MATERYAL VE METOD………. 21 4- BULGULAR……… 24 5- TARTIŞMA………. 29 6- SONUÇ………. 36 7- KAYNAKLAR………. 37
KISALTMALAR
AML: Akut myeloblastik lösemiALL: Akut Lenfoblastik lösemi APL: Akut promiyelositik lösemi TDP: Yaygın damar içi pıhtılaşma; PAS: Peryodik asit shift
EGIL: European Group for the Immunological Classification of Leukemias WHO: World Health Organizatıon
CBFβ: Core binding factor β ETO: Eight-twenty-one
MYH11: Myosin heavy chain gene RARα: Retinoic acid receptor α PML: Promyelocytic leukemia MLL: Mixed lineage leukemia HOX: Homeobox
FLT3: FMS-like tyrosine kinase 3 ITD: Internal tandem repeat
EVI1: Ecotropic Viral Integration Site 1
CEBPA: CCAAT enhancer binding protein alpha PLZF: Promyelositik lösemi zinc finger
NUP98: Nucleoporin 98 kDa
GATA1: GATA binding protein 1 (globin transcription factor1)) MPO: Miyeloperoksidaz
VEGF: Vasküler endotelyal growth faktör ATRA: All trans retinoik asit
ŞEKİLLER DİZİNİ Sayfa Şekil 1. t(15;17) sıklığı………25 Şekil 2. t(9;22) sıklığı………..25 Şekil 3. t(8;21) sıklığı………..26 Şekil 4. inv16 sıklığı……….26 TABLOLAR DİZİNİ TABLO 1. Akut miyeloid lösemide FAB sınıflaması……….. 3-4 TABLO 2. Akut lösemide sitolojik reaksiyonlar……… .4
TABLO 3. Akut lösemi WHO sınıflaması………5-6 TABLO 4. AML gelişimine katkıda bulunan çevresel faktörler………….. …8
ÖZET
Giriş ve amaç: Akut miyeloid lösemi; hematopoetik öncül hücrelerin heterojen
klonal bir hastalığıdır ve sıklığı yaş ile artar. Klonal çoğalan öncül hücrelerin matür hücrelere farklılaşma yeteneği yoktur ve normal hematopoezi de baskılarlar. Kemik iliğinde blast sayısının %20’den fazla olması tanıyı koydurur. Mevcut WHO sınıflaması moleküler, sitogenetik ve önceki hematolojik hastalığa göre yapılır. AML’li olgularda 100’den fazla genetik bozukluklar tanımlanmış olmakla birlikte sıklıkla saptanan kromozomal bozukluklar t(8;21), t(15;17), inv(16), t(16;16)’dır. t(8;21), inv(16) ve t(15;17) kromozomal bozukluklar nispeten olumlu prognostik özellik gösterirler. t(9;22) ise kötü prognostik özellik gösterir. Biz bu çalışmada yeni tanı almış AML hastalarında t(8;21), inv16, t(15;17) ve t(9;22) sıklığını ve prognoza etkisini araştırdık.
Materyal Metod: Dicle Üniversitesi Tıp fakültesi Hematoloji servisine başvuran ve
yapılan tetkiklerde Akut Miyeloid Lösemi tanısı alan 82 hastanın t(15;17), t(8;21), t(9;22), inv(16) translokasyonları Real Time PCR yöntemiyle (Roche-lightCycler) çalışıldı.
Bulgular: Yaptığımız çalışmada; t(8;21) sıklığı %6, t(15,17) sıklığı %17, inv(16)
sıklığı %9,7 olarak saptanırken, t(9;22) hiçbir olguda saptanmadı. t(15;17) pozitif hasta grubundaki hastaların tümü ATRA tedavisi aldı ve bütün hastalar remisyona girdi, bu grupta relaps gözlenmedi. İzlem boyunca tüm hastalarda t(15;17) negatifleşti. t(8;21) pozitif hasta grubu ile negatif hasta grubu arasında relaps gelişim açısından fark bulundu. Tedaviye refrakterlik, toplam sağ kalım oranı, toplam sağ kalım süresi ve ölüm oranı açısından pozitif ve negatif grup arasında fark bulunmadı. İnv16 pozitif hasta grubu ile negatif hasta grubu arasında relaps sıklığı, tedaviye refrakterlik, toplam sağ kalım oranı, toplam sağ kalım süresi ve ölüm oranı açısından fark bulunmadı. Olumlu prognoza sahip hasta grubunda (t(8;21) ve inv(16)) relaps gelişim açısından negatif gruba kıyasla fark bulundu. Tedaviye refrakterlik, toplam sağ kalım oranı, toplam sağ kalım süresi ve ölüm oranı açısından fark saptanmadı.
Sonuç: AML’de translokasyonların varlığı prognozu etkilediği için mutlaka yeni tanı
konulan her AML hastasında translokasyonlarının saptanması gerekmektedir. Ayrıca her AML hastasının ek kromozomal anomalileri saptanmalıdır. Böylece tedavi seçeneği ve prognoz hakkında daha doğru bilgiler edinilebilir. Ek kromozomal anomalilerinin varlığını da gösteren daha büyük serili çalışmalar konunun daha iyi anlaşılmasını sağlayabilir.
SUMMARY
Background and Aim: Acute myeloid leukemia (AML) is a heterogenous clonal
malignancy of haematopoietic stem cells, has a increased incidence with age. Differentiation capability is being lost in these growing stem cell clone, and resulted with depressed normal haematopoiesis. The diagnosis based on detection of increased immatur blastic cells in bone marrow higher than 20%. Molecular, cytogenetic and previous haematologic disease constitute the main of the WHO classification. More than a hundered cytogenetic defect has been determined. However, the most often chromosomal abnormalities were t(8;21), t(15;17), inv(16) and t(16;16). Patients with t(8;21), inv(16) or t(15;17) have relatively good, and with t(9;22) have relatively poor prognosis. In this presented study, we investigate the incidence and impact of these most common chromosomal abnormalities on prognosis of patients with newly diagnosed AML.
Material and Methods: Totally 82 consecutive patients with AML who admitted to
Dicle University, Faculty of Medicine, Department of Haematology were included into the study. These most common chromosomal abnormalities, t(8;21), t(15;17), inv(16) and t(9;22) were studied with Real Time PCR (Roche-lightCycler).
Findings: In our study, the incidence of t(8;21) was 6%, of t(15,17) was 17%, of
inv(16) was 9.7%. t(9;22) was not detected in any of patients with AML. All patients with t(15;17) positivity were treated with ATRA-Ida protocole. Remission was achieved in all of them, and any of them did not relapsed. t(15;17) was disappeared in all of these patients during follow up period. A significant difference was found between t(8;21) positive and negative groups in terms of relapse. We did not find any difference between t(8;21) positive and negative groups in terms of refractoriness, overall response and overall survey. Similarly, there was no statistically significant difference in relaps rate, refractoriness, overall response and survey of patients with inv16 positive and negative patients. Patients with good prognostic chromosomal abnormalities (patients with t(8;21) or inv(16)) were significantly different from others in terms of development of relapse. However, there were no statistically significant difference in terms of refractoriness, overall response and survey between these groups.
Conclusion: Chromosomal abnormalities which effect the prognosis of the disease
should be investigated in all newly diagnosed AML cases. In addition, complete caryotypical analysis should be performed to determine additional chromosomal abnormalities, and to consider different treatment options, and thought about prognosis. Larger scale studies needed to determine the importance of additional chromosomal abnormalities.
1-GİRİŞ VE AMAÇ
Akut miyeloid lösemi; hematopoetik öncül hücrelerin heterojen klonal bir hastalığıdır ve sıklığı yaş ile artar. Klonal çoğalan öncül hücrelerin matür hücrelere farklılaşma yeteneği yoktur ve normal hematopoezi de baskılarlar. Kemik iliğinde blast sayısının %20’den fazla olması tanıyı koydurur. Mevcut WHO sınıflaması moleküler, sitogenetik ve önceki hematolojik hastalığa göre yapılır.
AML’li olgularda 100’den fazla genetik bozukluklar tanımlanmış olmakla birlikte, sıklıkla saptanan kromozomal bozukluklar t(8;21), t(15;17), inv(16), t(16;16)’dır. Bu kromozomal bozukluklar ve bunun varyantları yaklaşık olarak AML olgularının %40’ını oluştururlar. Tanımlanmış olan diğer kromozomal anomaliler ise olguların %10’undan azını kapsarlar. Geri kalan %50 olguda ise normal karyotip saptanmakta veya kromozomal anomali saptanamamaktadır. t(8;21), inv(16) ve t(15;17) kromozomal bozukluklar nispeten olumlu prognostik özellik gösterirler. t(6;9), 5(q), 7(q), t(9;22) ve kompleks sitogenetik anomaliler ise kötü prognostik özellik gösterirler. Bariz sitogenetik anomali göstermeyen AML’ler orta derecede prognoza sahip hastalıklar olarak ele alınırlar. Moleküler analiz tekniklerinin gelişmesi lösemiye dönüşüm mekanizmalarının daha iyi anlaşılmasını sağlamış ve yeni tanısal gösterge olmaya başlamıştır.
Akut miyeloid lösemili hastalar tedavi edilmedikleri takdirde infeksiyon ve kanamadan ölmektedir. Günümüzde; AML’de ortaya çıkan moleküler genetik değişikliklerin saptanması ve bunların tedaviyi yönlendirmedeki değerlerinin anlaşılması ile AML’li hastalarda saptanan moleküler anormalliklerle birlikte komorbid faktörler birlikte değerlendirilip tedavi stratejileri geliştirilmiştir.
İyi, orta ve kötü sitogenetik risk grubları kemoterapi ve kök hücre nakline yanıtı belirtir. Standart remisyon indüksiyon tedavisi antrasiklinlerle sitarabin kombinasyonudur. Remisyon sonrası tedavi; iyi sitogenetik riskli grup için 3-4 siklus yüksek doz sitarabin, orta ve kötü sitogenetikli grup için allojenik veya otolog kök hücre naklidir. Son yıllarda araştırmalar akut miyeloid löseminin hedefe yönelik tedavisine odaklanmıştır.
Akut promiyelositik lösemide, gerek klinik özelliklerindeki farklılıklar ve gerekse tedavisinde son 20 yılda sağlanan gelişmeler nedeniyle diğer AML tiplerinden farklı bir kategoride ele alınmalıdır. Son yıllarda tedaviye
all-trans-retinoik-asid (ATRA)’nın girmesiyle ve efektif kullanımıyla Akut promiyelositik lösemi kür şansının en yüksek olduğu AML alt tipi haline gelmiştir.
Bu çalışma, Dicle Üniversitesi Tıp fakültesi Hematoloji servisine başvuran ve yapılan tetkiklerde Akut Miyeloid Lösemi tanısı konulan hastalarda yapılmıştır. Çalışmada hastalarda t(8;21), t(15;17), inv(16), t(9;22)’nın sıklığı araştırıldı. Ayrıca translokasyonları olan gruplar ile olmayanlar prognoz açısından karşılaştırıldı.
2-GENEL BİLGİLER AKUT MİYELOİD LÖSEMİ
Akut lösemiler, lösemik hücrelerin farklılaşma ve olgunlaşma kusuru göstermeleri, normal kan hücrelerinin yapılamaması ve aşırı çoğalma kabiliyetinde olan lösemik hücrelerin, önce kemik iliğini ve ardından periferik kanı ve diğer dokuları istila etmesiyle ile karakterizedir (1). Akut miyeloid lösemi (AML); hematopoetik öncül hücrelerin (blast) heterojen klonal hastalığıdır (2). AML’de malign hücre, sürekli miyeloid ve monositik farklılaşma gösteren bir blasttır, hastaların yaklaşık olarak % 5 ile % 10’unda blastlar eritroid veya megakaryositik farklılaşmaya sahiptir; bu nedenle AKUT NONLENFOSİTİK LÖSEMİ daha net bir terim olarak düşünülmektedir fakat AML daha yaygın ve tavsiye edilen bir terimdir (3). Akut lösemiler, uzun yıllar onu tanımlayan müelliflerin isimleri ile veya kaynaklandıkları hücre dizesine göre sınıflandırılmıştır. 1976 yılında Fransız, Amerikalı ve İngiliz bir grup hematopatolog tarafından orijinal (French-American-British-FAB) sınıflaması (4) oluşturulmuş ve aynı sınıflama 1985 yılında yeniden gözden geçirilmiştir (5). FAB sınıflamasında akut lösemiler, morfolojik ve sitokimyasal boyanma özelliklerine göre gruplandırılmakta ve akut miyeloid lösemi (AML) ve akut lenfoblastik lösemi (ALL) olmak üzere 2 gruba ayrılmaktadır (6,7).
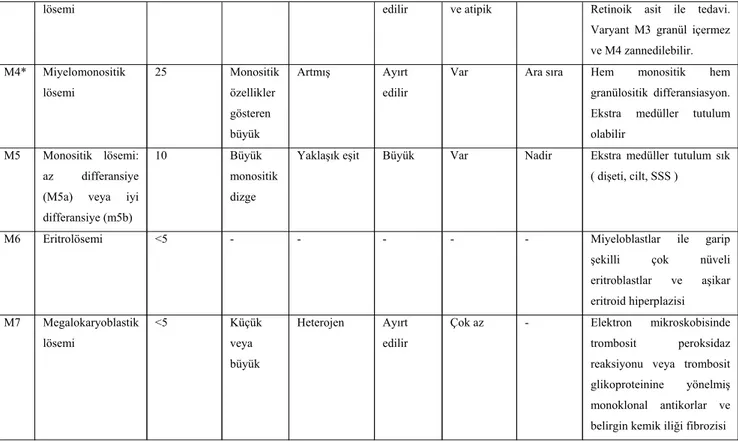
TABLO 1 : AKUT MİYELOİD LÖSEMİDE FAB SINIFLAMASI
Alt tip Morfoloji Erişkinlerde insidans (%) Hücre boyutu Sitoplazmanın nükleusa oranı Nükleol Sitoplazmik granül Auer Çubukları Yorum M0 Matürasyonsuz Miyeloblastik lösemi <5 Küçük veya orta Eşit veya azalmış Genellikle ayırt edilir.
Yok Yok ALL ile karışır, doğrulanması için CD13 ve CD33 immün belirteçleri gerekir. M1 Minimal matürasyonlu miyeloblastik lösemi 15 Küçük veya büyük
Yaklaşık eşit Ayırt edilir
Çok az Ara sıra Özel inceleme yapılmazsa M7 veya L2 ile karışır: inv (3) trombositozla beraberdir. M2 Matürasyonlu
miyeloblastik lösemi
25 Büyük Yaklaşık eşit Ayırt edilir
Var Ara sıra Ön plandaki hücreler blast
veya erken promiyelositlerdir. Extra
medüller tutulum veya splenomegali olabilir
KISALTMALAR: YDP: yaygın damar içi pıhtılaşma; *M4Eo: M4’ün bir alt tipidir ve kemik
iliğinde anormal eozinofillerle karakterizedir. Bu eozinofiller büyük bazofilik granüllere, tek loblu nüvelere veya pozitif periodic acid- Schiff (PAS) ve chloraacetate ( normal eozinofiller negatiftir ) boyanmasına sahiptir. Ekstramedüller tutulum sık olarak ortaya çıkar ve bu alt tip genellikle daha iyi
bir prognoza sahiptir.
Kaynak: Özkalemkaş F. Akut lösemiler. In: İç Hastalıkları, 2005. p.576-80
TABLO 2 Akut lösemide sitolojik reaksiyonlar
Kaynak: Özkalemkaş F. Akut lösemiler.In: İç Hastalıkları, 2005. p.576-80
lösemi edilir ve atipik Retinoik asit ile tedavi.
Varyant M3 granül içermez ve M4 zannedilebilir. M4* Miyelomonositik lösemi 25 Monositik özellikler gösteren büyük Artmış Ayırt edilir
Var Ara sıra Hem monositik hem granülositik differansiasyon. Ekstra medüller tutulum olabilir
M5 Monositik lösemi: az differansiye (M5a) veya iyi differansiye (m5b)
10 Büyük monositik dizge
Yaklaşık eşit Büyük Var Nadir Ekstra medüller tutulum sık ( dişeti, cilt, SSS )
M6 Eritrolösemi <5 - - - Miyeloblastlar ile garip
şekilli çok nüveli eritroblastlar ve aşikar eritroid hiperplazisi M7 Megalokaryoblastik lösemi <5 Küçük veya büyük Heterojen Ayırt edilir
Çok az - Elektron mikroskobisinde trombosit peroksidaz reaksiyonu veya trombosit glikoproteinine yönelmiş monoklonal antikorlar ve belirgin kemik iliği fibrozisi
REAKSİYON Yok veya zayıf
reaksiyon Orta derecede reaksiyon Kuvvetli reaksiyon Nonspesifik esteraz M0, M1 M2, M3 M4, M5 Florid inhibitörlü nonspesifik esteraz M0, M1, M5 M2, M3, M4 - Miyeloperoksidaz veya Sudan Black M0, M5, M7 M1, M4 M2, M3 Lizozim ( muramidaz ) M0, M1, M2, M3 M4 M5
FAB sınıflaması; immünfenotipleme, elektron mikroskobu, sitogenetik, moleküler biyolojik teknik yöntemlerini ve nadir lösemi tiplerini içermeyen bir sınıflamadır (6-7). Akut lösemide, hücre yüzey antijenlerinin ve sitogenetiğin öneminin fark edilmesi ve FAB sınıflamasının yeterli olmadığının gözlenmesi ile, akut löseminin bir çok özelliğini bir arada bulunduran sınıflamalar (MIC ve EGIL sınıflamaları) gündeme gelmiştir (8-9). MIC sınıflamasında akut lösemiler, hücre morfolojisi, immünfenotip ve sitogenetik özelliklere göre sınıflandırılmaya çalışılmıştır. EGIL sınıflamasında (European Group for the Immunological Classification of Leukemias) ise, AML’ler 2 veya daha fazla miyeloid hücre yüzey antijeni (MPO, CD13, CD33, CDw65, CD117) mevcudiyeti ile tanımlanmıştır (10-11).
Akut lösemide sitogenetik ve moleküler genetik anomalilerin bariz bir hale gelmesi ve bunların prognostik önem göstermelerinin belirlenmesiyle yeni bir sınıflandırma ihtiyaçı olduğu gözlenmiş ve 2001 yılında Dünya Sağlık Örgütü (World Health Organizatıon, WHO) tarafından akut lösemiler de dahil olmak üzere hemopoietik ve lenfoid neoplazmaları içeren yeni bir sınıflama yapılmıştır. Akut lösemi WHO sınıflamasında; morfoloji, immünfenotipleme, sitogenetik ve moleküler biyolojik özellikler göz önüne alınmış ve akut lösemi tanısı için kemik iliğinde blastik hücre sayısı % 30’dan % 20’ye indirilmiş ve aynı zamanda nadir lösemi tipleri de dahil edilmiştir (6). WHO sınıflamasında akut lösemiler; miyeloid, lenfoid ve serisi belirlenemeyen olmak üzere 3 gruba ayrılmıştır. Akut miyeloid lösemiler: a. Tekrarlayan sitogenetik anomalilerle seyreden AML, b. Çoğul seri displazi ile seyreden AML, c. Tedaviye ikincil AML ve MDS, d. Tanımlanan gruplara girmeyen AML olmak üzere dört gruba ayrılarak değerlendirilmiştir.
TABLO 3.
Akut lösemi WHO sınıflaması
Akut miyeloid lösemi
1. Tekrarlayan Genetik Anomalilerle Seyreden AML - t(8;21)(q22;q22), ( AML 1/ETO ) ile AML
- Akut promiyelositer lösemi (t(15;17)(q22;q12 ), (PML/RAR-alfa) ile AML) ve varyantları
- 11q23 (MLL) anomalisi ile AML 2. Çoğul seri displazi ile seyreden AML
- Önceden miyelodisplastik sendromlu (MDS) - Önceden miyelodisplastik sendrom olmadan 3. Tedaviye ikincil AML ve MDS
- Alkilleyici ajanlarla ilişkili
- Topoizomeraz II inhibitörleri ile ilişkili 4. Tanımlanan gruplara girmeyen AML - Minimal farklılaşma gösteren AML
- Olgunlaşma göstermeyen akut miyeloblastik - Akut miyelofibrozis ile panmiyeloz lösemi
- Granülositik olgunlaşma gösteren akut miyeloblastik lösemi - RAR alfa rearrajmanı göstermeyen akut promiyelositer lösemi - Akut miyelomonositik lösemi
- Akut monoblastik ve monositer lösemi - Akut eritrolösemi
- Akut megakaryoblastik lösemi - Akut bazofilik lösemi
- Miyeloid sarkom Akut lenfoblastik lösemi
1. Prekürsör B – lenfoblastik lösemi/lenfoma 2. Prekürsör T – lenfoblastik lösemi/lenfoma 3. Burkitt lenfoma / lösemi
Serisi belirsiz Akut lösemi 1. Bifenotipik akut lösemi 2. Farklılaşmamış akut lösemi
Kaynak: Head DR . Classification and differentiation of the acute Leukemias. In: Wintrobe’s Clinical Hematology, 2004.p.2063-76 (12-13).
AML’li olgularda 100’den fazla genetik bozukluklar tanımlanmış olmakla birlikte, sıklıkla saptanan kromozomal bozukluklar t(8;21), t(15;17), inv(16), t (9;11) ve t(16;16)’dır. Bu kromozomal bozukluklar ve bunun varyantları yaklaşık olarak AML olgularının %40’ını oluştururlar. Tanımlanmış olan diğer kromozomal anomaliler ise olguların %10’undan azını kapsarlar. Geri kalan %50 olguda ise normal karyotip saptanmakta ve kromozomal anomali saptanmamaktadır (14-16). inv16, t(8;21) ve t(15;17) kromozomal bozukluklar, CBFβ/MYH11, AML1/ETO ve PML/RARα füzyon protein yapımı ile suçlanmakta ve nispeten olumlu prognostik özellik gösterirler (17). t(6;9) veya 5(q), 7(q) kromozomal delesyonları ise, kötü prognoz özelliği oluştururlar (17). Bariz sitogenetik anomali göstermeyen AML’ler orta derecede prognoza sahip hastalıklar olarak ele alınırlar (16). Moleküler analiz tekniklerinin gelişmesi lösemiye dönüşüm mekanizmalarının nispeten daha iyi anlaşılmasını sağlamış ve akut lösemide yeni tanısal gösterge olmaya başlamışlardır. Örneğin AML’de internal FLT3-ITD mutasyonunun veya EVI1 sinyal faktöründe artmış mRNA ekspresyonunun belirlenmesi kötü prognozu işaret ederken, CEBPA transkript faktöründe mutasyonun saptanması iyi prognozu göstermektedir (18). Ancak hala AML’li olguların yarısında sitogenetik anomali veya moleküler özellik saptanabilmiş değildir (16,18).
Epidemiyoloji Etyoloji
Epidemiyolojik çalışmalardaki bulgular AML patogenezinde, çevresel, mesleksel ve genetik faktörlerin rol oynadığını ortaya koymuştur (19,20,21). İnsidans oranları gelişmiş ülkeler ve endüstrileşmiş şehirlerde daha fazladır. AML en sık görülen lösemidir ve olasılığı 40 yaşından itibaren yaşla artar, prevalansı 100 bin kişide 3,8’den, yaş ile 100 binde 17,9’a artar, erkeklerde bayanlara göre daha sıktır (3:2) (22). Genetik faktörler de AML patogenezinde yer almaktadır (23). Bu faktörler konjenital defektler (Down sendromu, Bloom sendromu, Monosomy 7 syndromu, Klinefelter sendromu, Turner sendromu, Neurofibromatosis, Conjenital dismorfik sendrom) ve kemik iliği yetmezlik sendromları (Fanconi anemisi, Dyskeratosis congenita, Shwachman-diamond sendromu, Amegakaryocytic thrombocytopenia, Blackfan-diamond sendromu, Kostmann agranulocytosis, Familial aplastic anemia) olarak ayrılabilmektedir(24). Çevresel faktörlerde AML patogenezinde yer almaktadır ( 25 ).
TABLO 4.
AML GELİŞİMİNE KATKIDA BULUNAN ÇEVRESEL FAKTÖRLER:
Solventler ( benzene ) Sigara içimi
İyonize radyasyon
Atom bombasına maruziyet Nükleer reaktörlere maruziyet Medikal radyasyon Noniyonize radyasyon ( ? ) Kemoterapi Alkilleyici ajanlar Topoizomeraz II inhibitörleri Başka ilaçlar Kloramfenikol Fenilbutazon
AML; kauçuk, boya, mumyalama sıvıları, böcek zehirleri, etilen oksit, gazolin, kümes hayvanları ve elektrik kablolarına maruz kalan işçilerde rapor edilmiştir (20,26).
KLİNİK PREZANTASYON
AML’li hastalarda semptomlar ani başlar ve anemiye özgü semptomlar ön plandadır (2,27). En yaygın şikayet nonspesifik yorgunluk hali, kırgınlık ve keyifsizlik şeklindedir. Hastaların % 15-20’sinde ateş mevcut bir özelliktir. Peteşi, epistaksis ve vücutta morarmalar tanıda hastaların yarısında görülen bulgu ve semptomlardır. Hastaların % 20’sinden azında kemik ağrısı görülmektedir. AML hastalarının yarıdan fazlasında organomegali ve adenopati bulunmasına rağmen bu bulgular ALL de daha yaygındır. Lösemik cilt infiltrasyonları hastalığın seyri boyunca hastaların % 13’ünde meydana gelir ve diğer ektramedüller alanların tutulumu ile ilişkilidir (28). Diş eti hiperplazisi akut monositik lösemide görülür. AML ile ilişkili benign cilt lezyonları Sweet sendromu ve piyoderma gangrenosumu içerir ve bu lezyonlar ağrılıdır ve steroide cevap verirler (29). Tanıda santral sinir
sistemi tutulumunu belirlemek, lumbal ponksiyon daima yapılmadığından, zordur (30-35). Yetişkinlerin %16’sında meningeal hastalık rapor edilmiştir (35). İntrakraniyal kitleler nadiren lösemik menenjitler ile bir aradadır ama intrakraniyal kitleler inv (16) ile ilişkili FAB M4Eo de rapor edilmiştir (32). Profilaktik SSS tedavisi AML’li hastalarda rutin olarak verilmemelidir ancak bazı klinisyenler özellikle çocuklarda ve AMoL hastalarda yada beyaz küre sayısı 100.000 den fazla olan hastalarda profilaksiyi savunurlar (34). Miyeloid sarkom AML’li vakaların % 2 ila %14’ünde meydana gelen ekstramedüller bir tümördür (36-41). Bu tömürlerin bazısı yeşil göründüğünden dolayı chloroma diye adlandırılır (39-41). Eğer Auer çubukları saptanmış ya da miyeloid orijin immünohistokimyasal ve sitokimyasal metodlarla doğrulanmışsa tanı konulabilir (39). Tümörler genellikle lokalizedir, sık sık kemik, periosteom, yumuşak dokular, lenf nodları ve cildi tutar. Miyeloid sarkom radyosensitif olmasına rağmen bir çok olguda sistemik kemoterapi verilmiştir (39). Testiküler tutulum AML’de ALL’den daha azdır ve testiküler tutulumun tedavisi bilateral testiküler radyasyon ve sistemik kemoterapidir (41-43). Pulmoner semptomlar, trombositopeniden dolayı hemorajili ya da nötropeniye bağlı infeksiyonlu ve lökostazlı hastalarda meydana gelir (41). Gastrointestinal semptomlar özellikle tifilit ve perirektal abse infeksiyonlarını içerir, Tifilit tedavisi nazogastrik sonda ve antibiyotikli medikal destektir (44-45). Obstrüktif sarılık granülositik sarkoma sekonder olarak gelişir ve AML’li hastalarda nadiren hepatik yetmezlik mevcuttur (41).
LABORATUAR BULGULARI
AML’li hastalarda kan değerleri geniş çaplı farklılıklar gösterir (46). Hastaların yarıdan fazlasında lökosit sayısı yüksektir ama % 20’sinden azında lökosit sayısı 100.000’den yüksektir. Blastlar genellikle periferik yaymada tanınırlar. Sitopeniler, hematopoetik yetersizlikten kaynaklanır ve semptom ve bulgulara katkı sağlar. AML’de anemi baskın olarak normokrom normositerdir, çekirdekli eritrositler ve retikülositopeni mevcuttur. Tanıda ağır seyredebilecek trombositopeni DİC’e eşlik edebilir. AML’de DİC, ALL’den daha yaygındır; en çok, ilk kez Hıllstad tarafından tanımlanan akut promiyelositik lösemide görülür (47). AML’li hastaların %50’sinde hiperürisemi bulunmuştur (48). Serum laktat dehidrogenaz özellikle
monositik subtiplerde (M4/M5) yükselmiş olabilir (49). AML’li hastalarda hiperkalsemi olabilir ama hipokalsemi daha yaygındır (50).
Hiperlökositoz 100.000’den fazla kan blast sayısıdır, monositik komponentsiz AML’de nadirdir ve AMoL ile AMMoL de daha yaygındır (40, 51, 52). Pulmoner lökostaz belirtileri dispne, taşipne, intertisyel infiltrasyon, solunum yetmezliğidir ve kötü bir prognoz ile ilişkilidir (53- 55). Santral sinir sistemi lökostaz semptomları başağrısı ve bulanık görmedir. Fatal intraserebral hemoraji meydana gelebilir (30). Priapizm, büyümüş böbrekler, hiperürisemi ve renal yetmezlik genitoüriner tutulum özellikleri olarak görülmektedir (41). Hiperlökositoz; santral sinir sistemi hemorajisi, solunum yetmezliği, SSS tutulumu ve daha yüksek relaps oranından dolayı kötü prognoz ile ilişkilidir (30,56,57,58). Hiperlökositozlu hastalarda lökostazın riskini azaltacak tedavi seçenekleri, lökoferez, yüksek dozda hidroksiüre verme ve acilen indüksiyon kemoterapisinin verilmesini içerir (40,52,58).
BİYOLOJİK ÖZELLİKLER
Akut Miyeloid Lösemi, olgunlaşmamış veya blast hücrelerinden oluşan kemik iliğine bağlı bir neoplazmdır.
Nonlenfoid farklılaşma morfolojik, sitokimyasal ve immunolojik çalışmalar ve elektron mikroskobu ile kanıtlanabilmektedir (59-60). Sitogenetik analizler (61), izoenzim çalışmaları (62) ve sınırlı fragman uzunluk polimorfizm analizleri (63,64) AML’nin klonal yapısını incelemektedir.
AML’ de kromozomal translokasyonlar:
AML’de görülen translokasyonların çoğu kimerik füzyon gen ürünü oluşturur.
1.CBF-core binding faktör: 20’ye yakın translokasyon CBF genini hedef alır. CBF grubundaki genler birçok dokunun farklılaşmasında rol oynayan çeşitli hedef genlerle heterodimer kompleksi oluşturarak işlev görür. DNA’ya bağlanarak işlev gören alfa ve DNA’ya bağlanmadan transkripsiyonel aktiviteyi arttıran beta alt üniteleri vardır. Öncelikle t(8;21), AML1-ETO translokasyonu tespit edilmiş olup, buradaki AML1 geni CBF’nin DNA’ya bağlanarak transkripsiyonu başlatma özelliğine sahip alfa1 alt grubu üyesidir (65).
16. kromozomun inversiyonu- inv(16) sonucu fonksiyonu değişen diğer bir CBF’de, CBF betadır. Burada CBFβ-MYH11 füzyonu söz konusudur. Her iki translokasyon
da hematopoezisi bozsalar da tek başlarına AML başlatmaya yeterli değillerdir. AML1-ETO ve CBFB-MYH11 translokasyonları AML’li olguların yaklaşık olarak sırasıyla %2-12 ve %1-5 kadarında görülür ve her ikisi de iyi prognozla birliktelik gösterir. Bu nedenle hastalık tanısı ve takibinde kullanılan markırlardır (65).
2. RARα-Retinoik asit reseptör alfa:
17. kromozomda bulunan RARalfa ile 15. kromozomda lokalize PML geni ya da 11. kromozomda lokalize PLZF (promyelositik lösemi zinc finger) geni füzyonu oluşur. Bu durum fenotipik olarak gelişimin promiyelositik safhasında durmasına neden olur (65). PML-RARα füzyon proteini DNA’ya bağlanma ve dimerizasyon kapasitesini yitirmemesine rağmen retinoik aside bağlanabilirliği değişmekte ve hedef genlerin transkripsiyonu bozulmaktadır (66,67).
t(11;17)(q23;q21) translokasyonunda ise RARα geni 11.kromozomdaki PLZF geni ile birleşmektedir. Ortaya çıkan füzyon proteini RARα ya benzer şekilde RARα nın DNA’ya bağlanma kapasitesini değiştirmektedir (65). Ancak yine CBF mutasyonları gibi, RARα mutasyonlarının da lösemi gelişimi için başka mutasyonların katkısına gereksinimleri vardır (65).
3. Mixed Lineage Leukemia (MLL) geni translokasyonları:
11q23 bölgesini içeren translokasyonlar AML yanında ALL’de görülür. 30’dan fazla partner gen bu bölgede bulunan MLL geni ile translokasyon yapar. Bu durum 1 yaş altı infant lösemilerinde en sık görülen genetik değişikliktir. Ayrıca yine tedaviye bağlı sekonder lösemilerde de 11q23 aberasyonlarına sıklıkla rastlanır (65). AML’li hastaların yaklaşık %15’i 11q23 kromozomu içeren duplikasyon, delesyon, translokasyon gibi sitogenetik anormalilere sahiptir (68).
4. Homeotik genler-HOX :
HOX gen ailesine ait genler omurga gelişimi ve ayrıca normal hematopoez de önemli rol oynarlar. AML’de HOX genleri, çeşitli tipleriyle MLL geni ile translokasyon yapmaktadır. Burada HOX genlerindeki sorun, normal hematopoezisi regüle edici fonksiyonunu kaybetmeleri tarzında olmaktadır (65). HOX genlerini hedefleyen kromozomal translokasyonlar arasında t(7;11) NUP98-HOXA9 ve t(2;11) NUP98-HOXD13 sayılabilir (69).
AML de görülen nokta mutasyonları:
1. RAS gen ailesi mutasyonları: Onkogenik RAS mutasyonları AML ve MDS’de görülür. Tipik olarak N- ve K- RAS genlerinin 12, 13 ya da 61. kodonlarında olur. Bu mutasyonların sıklığı farklı çalışmalarda değişmekle beraber %25-44 arasındadır. Yapılan çalışmalarda aktive edici mutasyon varlığının kötü prognozla birlikte olduğu gösterilmiştir(65).
2. Reseptör tirozin kinazı aktive edici mutasyonlar:
AML olgularının oldukça yaygın bir bölümünde FLT3 ve c-KIT geninde aktive edici mutasyonların varlığı gösterilmiştir. FLT3 normal hücrelerde proliferasyon, farklılaşma ve hayatta kalımda önemli bir rol oynar (70). FLT3 genindeki mutasyonlar AML’li hastaların yaklaşık olarak %40’ında bulunur (71). Olguların yaklaşık %20-25’inde genin bir bölgesinin (juxtamembranedomain) duplikasyonu (internal tandem duplication) aktive olmasına neden olur. Bu mutasyonla genin otoinhibitör bölgesinin hasara uğradığı ve kinaz aktivitesinin ortaya çıktığı gösterilmiştir. Olguların yaklaşık %5-10’unda ise genin D835. pozisyon (aktive edici loop) bölgesinde oluşan nokta mutasyonu yine tirozin aktivitesine neden olmaktadır (65). FLT3 mutasyonları AML hastalarında kötü prognozla birliktelik gösterir (71). AML olgularının yaklaşık %5’inde c-KİT geninin D816 pozisyonu mutasyonları bildirilmektedir. Bu mutasyon da yine genin aktive edici loop bölgesinde olup genin kinaz aktivitesini arttırmaktadır (65).
3.AML1, CEBPA, GATA1 mutasyonları:
AML1 geni lösemilerde sıklıkla transloke olmakla birlikte, bazı kalıtsal AML olgularında da genin fonksiyon kaybına neden olan mutasyonlar gösterilmiştir. Bunun yanında olguların %3-5’lik bir bölümünde AML1 mutasyonları da tespit edilmiştir. Bunlar daha çok AML1 ve trizomi 21 ile birlikte görülen AML ya da MDS olgularıdır (65).
CEBPalfa geni normal miyeloid gelişimi için gerekli bir faktördür (72). CEBPα genindeki mutasyonlar daha çok M2 alt tipindeki olgularda bildirilmiştir ve son çalışmalar mutasyonun iyi prognozla ilişkisini göstermiştir (65).
GATA1 gen mutasyonları M7 ve Down sendromun da gelişen lösemilerde görülmektedir (65).
Klinikopatolojik sendromlar:
1. Minimal Olarak Farklılaşmış AML: Bu vakalardaki blastlar agranüler ve auer body içermezler. AMLMO tanısı MP0-pozitifliği ve Sudan Black B-pozitif hücrelerinin %3’ünden daha azını fakat miyeloid antijenleri ifade eden (CD13,33,117) lösemi hücrelerinin %20’sinden fazlasını gerektirmektedir (73). Lenfoid ilişkili antijenler (CD2,7,19) ve TdT yaygın olarak AML MO’da ifade edilir (73). Kromozom 8 ve 13 trizomileri yaygın şekilde görülmektedir (74). AMLMO daha yaşlı (ortalama 60 yaş) hastalarda meydana gelme eğilimindedir. AMLM0’de survival ve komplet remisyon oranı kötüdür (74).
2. Maturasyonsuz AML: AML (FAB M1) herhangi bir maturasyon kanıtı (<%10 promiyelositler veya diğerleri, daha olgun hücreler) olmaksızın miyeloblastların üstünlüğü (>%90 ) ile tanımlanmıştır. Blastlar (CD13,CD14 veya CD33) içeren miyeloid antijenleri expresse ederler. AMLM1 AML’li vakaların %10-20’sini oluşturur. Erişkinlerde çoçuklardan daha yaygındır (75). AMLM1’e eşlik eden herhangi bir spesifik sitogenetik markır mevcut değildir.
3. t(8;21) ve Maturasyonlu AML: t(8;21)(q22;q22) AMLM2 vakalarının %29-40’ında tespit edilmiştir. Klinik özellikler splenomegali, kloroma ve tanıda anemi ve trombositopeniyi içerir. İmmünofenotipik markırlar miyeloid antijenlerin görünümünü içerir. CD34, CD19, CD56 gösterilirken CD2 ve CD7 yoktur (76). t(8;21) li AML hücrelerinde CD56 antijeninin expresyonu daha kısa hastalıksız sağ kalım ile ilişkilidir (77).
4. Akut Promiyelositik lösemi: AML’li hastaların %5-10’unda görülür, t(15;17) translokasyonu ile karakterizedir (78). Hastalar genellikle ortalama 30-38 yaşlarında genç hastalardır (75). Hastaların %90’ından fazlasında DIC, sekonder hemorajik olaylar görülür (75). Lökopeni genellikle daha yaygın olan hipergranüler APL’de mevcuttur, buna karşın lökositoz mikrogranüler varyantlarda meydana gelme eğilimindedir (75). Lösemi hücreleri karakteristik olarak çok belirgin granüllere sahiptir, auer rodların destesiyle birlikte nükleusu anlaşılmaz hale getirebilir. Mikrogranüler varyant APL, APL vakalarının %20’sinde görülür ve ışık mikroskobuyla tanınamayan
ancak elektron mikroskobuyla saptanan küçük granüllere sahiptir (79). Miyeloid antijenler CD13 ve CD33 görülürken HLA-DR yoktur. Mikrogranüler varyant CD34 ve T cell antijen CD2 eksprese eder (80).
5. Anormal Eozinofilli Akut Miyelomonositik lösemi ve kromozom 16’nın inversiyonu: AML’li hastaların yaklaşık olarak %5-10’unda kemik iliğinde miyeloblasttik/monoblastik bir infiltrasyon ve monositoz görülür. Ortalama yaş 45-50’dir. Organomegali yaygındır, lökositoz hastaların çoğunda görülür (32). Santral sinir sistemi hastalıkları ve lösemia cutis yaygındır. Tüm AMLM4Eo’de CD13 ve CD34 exprese edilir (81). CBF-β-MYH11 füzyon transkripsiyonu AMLM4Eo de RT-PCR ile tanınabilir ama eozinofilisiz AML M4’lü hastaların yaklaşık %10’unda saptanabilir (82). Komplet remisyon oranları diğer lösemi tiplerinden daha yüksektir, hastalıksız yaşam süresi iyidir ve bu nedenle kemik iliği transplantasyonu ilk remisyondan ziyade relapsdan sonra düşünülmelidir (82). İnv16’lı AML hastaları daha yüksek santral sinir sistemi nüksüne sahiptir (83).
6. Akut monositik lösemi: AMoL, AML vakalarının %2-10’unu izah eder (84). M5, M5a (monoblastları içeren monositik hücreler %80’den fazla) ve M5b (iyi farklılaşmış, %80 monositik dominant olarak promonosit ve monosit) diye ayrılmıştır (85). Monoblastlar CD68’e ve lizozime antikorlarla boyanarak tanınabilir (75). Ekstramedüller hastalık diğer tiplere oranla daha yaygındır, kutanöz lezyonlar, diş eti hiperplazisi ve santral sinir sistemi hastalığını içerir (86). AMoL kötü prognostik faktörlere eşlik ettiği ve daha kısa yanıt süresine sahip olduğu öne sürülmektedir (86)
7. Eritrolösemi: Eritrolösemi AML olgularının %5’inden azını izah eder ve eritroid/miyeloid oranı noneritroid popülasyonda %20 blast ve eritroid popülasyonun %50 ya da daha fazlası eritroid öncülleridir. Eritrolösemi genellikle 50 yaş ve üzerindeki hastalarda yaygın olarak erkeklerde meydana gelir (85). Aneuploidy hastaların neredeyse üçte ikisinde görülür (87). Hastaların üçte birinde kemik ağrısı majör semptom olabilir (85). Hipergamaglobülinemi, antinükleer antikor, romatoid faktör, coombs testi kemik ağrılı hastalarda bulunabilir (88). Diğer tip AML’lere göre daha kötü prognozludur.
8. Akut megakaryositik lösemi: AML vakalarının %5-10’unda görülür (85). CD13 ve CD33 pozitif iken CD34 yoktur. Tanı lösemik hücrelerde en az bir platelet antijeninin ekspresyonuna bağlıdır (CD41, CD42b, CD61 ve faktör VIII related antijen). Kemik iliği aspirasyonu önemli fibrozisden dolayı zor olabilir. Sitogenetik anomaliler heterojendir ama kromozom 8 ve 21’in anomalilerini içerir (85). Osteosklerotik ve osteolitik lezyonlar radyografik olarak gösterilmiştir (85). Sitopeniler genellikle görülür ama hastaların yaklaşık %30’u yüzbini aşan trombosit sayılarına sahiptir (85).
Başka klinikopatolojik sendromlar.
Akut bazofilik lösemi: Çok nadirdir, kutanöz hastalık, litik kemik lezyonları, hiperhistaminemi semptomları görülebilir. Karakteristik sitokimyasal boyaları, toluidin mavili metakromatofilik pozitiflik ve asitfosfataz pozitifliğidir (89).
Akut panmiyelosis: AMLM7 bölümünde not edilen akut miyelofibroz ve akut myelosklerozun yerini almıştır (90). Çok nadirdir, pansitopeni mevcut olup splenomegali nadirdir (89).
Prognoz:
Klinik özellikler, morfoloji, yüzey markırları ve sitogenetikler AML’deki klinikopatolojik sendromları tanımlamak için kombine edilir ve prognoz genellikle spesifik faktörlerin kombinasyonuyla belirlenir (91,92). Kötü prognoza sahip yaşlıca ve daha genç (infantlar<2 yaş) hastalarla birlikte yaş önemli prognostik faktördür (93). Hastanın klinik durumu, sonuçları etkiler (94). MDS veya miyeloproliferatif sendrom gibi kemik iliği hastalıklarını daha önce geçiren hastalar veya kemoterapi hikayesi olanlar, de novo AML’lilere kıyasla daha az bir sağkalıma sahiptir (95). Bazı serilerde hiperlökositoz kötü bir prognoz göstermesine rağmen AML’deki bu korelasyon ALL kadar güçlü değildir (96). Auer rodları içeren M1-M4 ve M2-M4Eo olumlu morfolojik özellik içerirken, eritrolösemi, akut megakaryositik lösemi, MDS’den gelişen AML olumsuz özellikler içerir (91,92). Prognozda yüzey markırlarının etkisi değişkendir ve diğer faktörler ile birbirlerine bağlıdırlar dahası antijen ekspresyonu relapsda sıklıkla değişir (97). Sitogenetik alt gruplar prognozu diğer herhangi bir faktörden daha iyi tanımlarlar (98). t(8;21), inv(16), t(15;17) varlığı olumlu olarak sayılmıştır. Kötü prognostik gruplar, kromozom 5, 7, 3q26 ve kompleks karyotip anomaliler ile MDS ve kemoterapi öyküsü olan hastaları içerir
(99). Normal bir karyotip genelde orta derece bir prognoza eşlik ederken ancak RT-PCR gibi moleküler teknikler prognostik öneme sahip olabilen (FLT-3 ve MLL gibi) normal karyotipli AML’de moleküler değişikliği tanıyabilir (100). MDR genlerinin ekspresyonu, AML’de advers prognostik markırlar olarak tanımlanmıştır (101). AML’de prognoz moleküler markırların değerlendirilmesiyle de belirlenebilir. Özellikle büyüme faktörlerini kodlayan genler ve onların reseptörleriyle ilişkilidir ve tanıda en iyi çalışılan reseptör FLT-3’tür (102). AML’li hastaların %20 ila %34’ünde PCR amflikasyonu ve jel elektroforez yoluyla saptanabilirler (102). FLT-3 gen mutasyonları; daha yüksek periferik blast sayımı, normal sitogenetikler, t(15;17) ve FAB M5 hastalıklarına eşlik eder, FLT-3 mutasyonlarının tam remisyon oranıyla ilişkisi olmamasına rağmen, nüksediş ve kötü sağkalımı önceden bildirir (102). EVI1 proteinin artmış ekspresyonu sadece 3q26 içeren AML’liler de değil aynı zamanda diğer kötü karyotipler ile ilişkilidir ve kötü prognoza sahiptir (103). Azalmış apoptozis ve artmış anjiogenesis, AML’de leukemogenesis ve kötü prognoza yardımcı olabilir. Artmış bcl-2 proteini expresyonu, lösemi hücreleri üzerinde bir antiapoptotik etki oluşturmaktadır ama bcl-2 expresyonunun bilinen prognostik faktörlerle ilişkisi olan çalışmalar karışık sonuçlar vermiştir (104). VEGF’nin aşırı expresyonu, artmış anjiogenezle ilişkili ve AML’de çok kötü bir yanıt ve sağkalıma eşlik etmiştir (105).
Tedavi:
AML tedavisi 2 kısma ayrılabilir, ilk kısım remisyona sokma (remisyon indüksiyon) ikinci kısım ise remisyon sonrası tedavidir (27). AML tedavisindeki ilk amaç hastayı tam remisyona sokmaktır (106). Tam remisyon; absolü nötrofil sayısının 1000/mm3, trombosit sayısının 100.000/mm3 üzerine çıkması, kemik iliğindeki blast sayısının %5’in altına inmesi olarak tanımlanır (107). Hastalarda öncelikle anemi ve infeksiyon tedavisi yapılmalı, lökostaz, tömür lizis sendromu bulguları varsa bu sorunlar acil olarak tedavi edilmeli ve hasta remisyona sokma tedavisine hazır hale getirilmelidir (106).
Remisyon indüksiyon tedavisi (Rİ): Standart tedavi 7+3 denilen Ara-C 100-200 mg/m2/gün devamlı i.v infüzyon 1-7 gün ve daunorubicin 45 mg/m2 veya 60 mg/m2 i.v 1-3 gün kemoterapi rejimi olup hastalarda %65-75 oranında tam remisyon sağlanır (107). Kemoterapi rejiminde daunorubicin yerine idarubicin, mitoxantron
gibi diğer antrasiklinleri kullanmanın, rejime etoposide, fludarabin, topetecan veya multidrug rezistan inhibitörü eklemenin standart 7+3 rejimine üstünlüğü gösterilememiştir (108). Remisyona sokma tedavisinin 10-15’ci gün yapılan kemik iliği aspirasyonunda blastlar mevcut ise remisyona sokma tedavisinin ikincisine aynı ilaçlarla başlamak birçok grup tarafından önerilmektedir (27). 10-15’ci gün yapılan kemik iliği aspirasyonunda hipoplazi mevcutsa 10-14 gün sonra kemik iliği aspirasyonu tekrarlanır ve bazen aspirasyonda rejenere olmakta olan kemik iliği bulgusu olarak %30-40 blast görülebilir, böyle bir durumda 1-2 hafta daha bekleyip kemik iliği aspirasyonu tekrarlanarak durum tekrar değerlendirilir (106).
Remisyon sonrası tedavi: Remisyona giren hastalarda remisyon sonrası tedaviler olarak; konsolidasyon kemoterapisi, otolog ya da allojenik kök hücre nakli tedavileri yapılabilir (107). Remisyon sonrası tedavi planı prognostik faktörlere bakılarak yapılır, en önemli prognostik faktör hastanın karyotipidir (107).
İyi prognoz karyotipi olan (t(8;21) ve inv(16)) ve standart tedavi ile remisyona giren hastalarda 4 siklus cytarabine 3g/m2 her 12 saatte bir vermek standart doz veya intermediate doz cytarabine vermekten daha faydalıdır (27). Yüksek doz cytarabine tedavisi; Core binding lösemi denen, t(8;21) ve inversiyon 16 mevcut hastalarda; standart remisyon sonrası tedavidir ve 3 veya 4 siklus cytarabine verilmelidir (106). Core binding lösemilerde otolog periferik kan veya allogeneik kök hücre transplantasyonunun yararı yoktur (106). Ancak CBL olup da Flt3 ya da CD56 pozitif hastalarda yüksek doz cytarabine tedavisi yetersiz kalmaktadır ve bu hastalarda allogeneik kök hücre nakli düşünülmelidir (106).
İntermediate riskli hastalarda; konsolidasyon kemoterapisini takiben otolog veya allogeneik kök hücre nakli önerilmektedir (106).
Kötü riskli AML’li hastalarda; HLA uyumlu kardeşden, akraba dışı donörden allogeneik kök hücre nakli önerilen tedavidir (109). Miyelodisplazi ya da hematolojik hastalık sonrası gelişen, 5q-, 7q-, tr:8, t(6;9), t(9;22) gibi kötü prognostik sitogenetik bulgusu olan AML’li hastalarda allogeneik kök hücre nakli standart tedavi olarak kabul edilmektedir (106). Donörü olmayanlarda otolog kök hücre nakli ya da yüksek doz Ara-C ile konsolidasyon önerilir (106). Allogeneik transplantasyon planlanan hastalara ise remisyona girme sonrası transplantasyon önerilir ve konsolidasyon tedavisi vermenin avantajı gösterilememiştir (110). Donörü olmayan
ve otolog transplantasyon planlanan hastalarda ise Rİ tedavisi sonrası konsolidasyon tedavisi verilmesi ve takiben otolog kök hücre nakli yapılması önerilir (111).
Relaps AML tedavisi; Hastaların %50’sinden fazlası relaps olur ve ortalama relaps süreleri 3 aydan 12 aya kadar değişir (112). Reindüksiyon kemoterapisine en iyi cevap göstergeleri yaş, karyotip, ilk remisyon süresidir (113). Relaps yapan hastalardaki cevap oranları kötü sitogenetik özelliklere sahip hastalarda %40’ın altında iken iyi karyotipik özelliklere sahip hastalarda %90’ın üzerindedir (114). İlk remisyon süresi 6 ayın üzerinde ise cevap oranı belirgin olarak daha yüksek olma egilimindedir (115). Relapslı hastalarda hastalık, en iyi kurtarma tedavisini belirlemek için sitogenetikler, yaş ve birinci tam remisyonun süresine göre sınıflandırılabilir (116). Yüksek risk AML için yeni randomize MRC çalışması ( MRC AML-HR); G-CSF ile kombine olsun ya da olmasın fludarabin+sitarabin kombinasyonun standart ADE (cytarabine, daunorubicin ve etoposid) kombinasyonuna üstün olmadığı gösterilmiştir (117).
Hedefe yönelik tedaviler:
Anti-CD33 Antikor: CD33 antijeni pluripotent hematopoietik kök hücreler tarafından eksprese edilmemesine karşın AML blastlarının yaklaşık %90’ı tarafından eksprese edilmektedir. Bu yüzden bu antijen, AML blastlarının antikor aracılıklı yıkımı için bir hedeftir. Gemtuzumab ozagamisin sitotoksin calicheamicin ile konjuge rekombinant anti CD33 monoklonal IgG4 antikorudur (118). Konjuge antikor hızlı bir şekilde hücre içine alınır ve ardından apopitoza sebep olur (119). Başka bir anti CD33 olan HuM195 minimal rezidüel hastalıklı hastalarda kullanılmış, lösemik tutulum bölgesine radyoaktif madde yaymak için I-131, yttrium-90 ve bismuth-213 ile konjuge edilmiştir (120).
Sinyal iletim yollarını hedefleyen tedaviler:
FLT3 inhibitörleri: Aktive FLT3 reseptör mutasyonları AML’li hastaların yaklaşık %30’unda saptanmıştır. PKC412, CT83518, CEP701 ve SU5416’yı içeren birkaç küçük molekül FLT3 tirozin kinaz inhibitörü tanımlanmıştır (121).
KIT tirozin kinaz inhibitörleri: Imatinib mesylate refrakter sekonder AML’de tam remisyon oluşturmuşsa da bu durum yaygın değildir (122).
Prenilasyon inhibitörleri:
Farnesyltransferaz inhibitörleri: RAS mutasyonu aktivasyonu AML hastalarının yaklaşık %15’inde görülmüştür (123). Farnesyltransferaz; hücre membranına RAS translokasyonu için gerekli olduğundan bu enzimin inhibitörlerinin RAS aktivitesini inhibe ettiği varsayılır (124). Birkaç farnesyltransferaz inhibitörü (BMS-214662, L-778123, R-115777: Tipifarnib, SCH-66336:Lonafarnib) AML hücre büyüme inhibitörü olarak çalışılmaktadır (125).
Geranylgeranyltransferaz inhibitörleri: Birçok protein farnesylasyona maruz kaldığından Geranylgeranyltransferaz-I inhibitörleri AML’de aktiviteye sahip olabilirler ve AML’li hastalarda farnesyltransferaz inhibitör monoterapisine resiztansı açıklayabilir (126). Statinler geranylasyonu inhibe ederler ve AML’nin lovastatine cevap veren vakaları bildirilmiştir (127).
Antianjiogenik ajanlar: Amifostin sitoprotektif etkili ve bir miktar antianjiogenik potansiyele sahip olduğu bilinmektedir, AML’de tam remisyona yol açabilir (123).
İlaç direncinin düzenlenmesi: P-glikoprotein, MDR-protein 1 ve göğüs kanser resiztans protein (BCRP) ekspresyonu tüm AML hastalarında bildirilmiştir (128). P-glikoprotein ekspresyonu daha kısa süreli remisyon ve azalmış remisyon oranları ile ilişkilidir (129).
Akut promiyelositik lösemi tedavisi:
Yeni tanılı APL hastaların tedavisinde ATRA(All trans retinoik asit)+Antrasiklin (İdarubisin 12mg/m2/gün 2, 4, 6 ve 8. günlerde) kombinasyonu standart rejim olarak benimsenmiştir (130). ATRA’nın en önemli yan etkisi ateş ve damar dışına sıvı sızmasının neden olduğu plevral-perikardiyal effüzyon, dispne ile giden retinoik asit sendromudur. Tedavisi 3-5 gün i.v 10mg günde 2 kez dexametazon ile tedavidir. 4-6 haftalık tedavi neticesinde, kemik iliğinde yeterli sellülalitenin ve normal maturasyonun saptanması, blast oranının %5’in altına inmesi, auer rod içeren hücrelerin kaybolması, çevresel kanda nötrofil sayısının 1500/mm3, trombosit sayısının 100.000/mm3 geçmesi ve bu tablonun en az 4 hafta devamlılığı tam remisyon olarak kabul edilebilir (131).
Konsolidasyon tedavisi: ATRA+Antrasiklin kombinasyonu ile remisyon elde olunduktan sonra 3 kür antrasiklin esaslı tedavi yöntemi hastaların %80-85’inde uzun süreli hastalıksız sağkalım ve potansiyel kür şansı yaratır (132).
İdame tedavisi: ATRA 45mg/m2/gün her 3 ayda bir 15 gün, 6-merkaptopürin 90mg/m2/gün devamlı ve haftada bir 20 mg/m2 metotreksat kullanan hastalarda 2 yıllık relaps oranının düşük olduğu bulunmuştur (133).
Relaps olan vakalarda ikinci remisyon indüksiyonu olarak ATO (Arsenik trioksit) önerilmektedir (132). ATO’nun lökositoz, hiperglisemi, EKG’de QT uzaması gibi hayatı tehdit edecek yan etkileri açısından dikkatli olunmalıdır (132).
Son zamanlarda relaps APL hastalarında bir monoklonal antikor olan gemtuzumab ozogamisin ve lipozomal ATRA’nında kullanılabileceğine dair yayınlar vardır (132).
3-MATERYAL METOD
Bu çalışma Nisan 2004 – Ocak 2008 tarihleri arasında Dicle Üniversitesi Tıp fakültesi Hematoloji servisine başvuran klinik, laboratuar, periferik yayma, kemik iliği sonuçlarıyla birlikte Akut Miyeloid Lösemi tanısı alan 82 hastada gerçekleştirilmiştir. Hastaların 41’i bayan, 41’i erkek olup, yaş ortalamaları 37,8’di. (15-76 yaş)
Çalışmaya katılan tüm hastaların fizik muayene bulguları, tam kan, biyokimya, periferik yayma, kemik iliği ve moleküler genetik sonuçları ile birlikte aldığı tedaviler çalışma formuna kaydedildi. Moleküler genetik verileri hematoloji laboratuarı tarafından aşağıda belirtildiği şekilde çalışıldı.
EDTA’lı tüpe alınmış tam kandan RNA izolasyonu (Roche-HighPure RNA isolation kit)
Lökosit eldesi (RBC Lizis Buffer): 1,5 ml’lik eppendorf tüpüne 1 ml RBC lizis buffer solüsyonu alındı, üzerine 500µL kan eklendi, elde karıştırıldı, 10 dakika karıştırıcıda oda sıcaklığında bekletildi. 2500 rpm’de 5 dakika santrifüj edildi. Dipte oluşan lökosit çökeltisine dokunmadan üsteki berrak kırmızı sıvı pipetle toplandı ve atıldı. Lökosit çökeltisi üzerine tekrar 1 ml RBC buffer eklendi, parmakla vurularak çökelti çözüldü. 2500 rpm’de 3 dakika santrifüj edildi. Tekrar üsteki sıvı pipetle atıldı. Kalan lökosit çökeltisinin üzerine 200µL pbs (fosfat buffer salin) eklendi ve çökelti süspanse hale getirildi. Böylece 200µL lökosit süspansiyonu elde edilmiş oldu.
RNA izolasyonu: Lökosit süspansiyonu üzerine lysis-binding buffer eklendi ve iyice karıştılırdı. Daha sonra bu karışım filtreli ve toplama kabı olan başka bir tüpe alındı. 10.000 rpm’de 15 saniye santrifüj edildi. Alttaki toplama tüpündeki sıvı döküldü, aynı tüp filtreli tüpe tekrar takıldı. Başka bir eppendorf tüpünde 90µL DNAaz buffer ile 10µL DNAaz 1 karışımı hazırlandı ve bu karışım filtreli tüpe eklendi. 15 dakika oda sıcaklığında bekletildikten sonra 500µL wash-buffer-1 eklendi, 10.000 rpm de 15 saniye santrifüj edildi. Toplama tüpündeki sıvı döküldü. Tüp tekrar filtreli tüpe takıldı ve 500µL wash-buffer-2 eklendi. 10.000 rpm’de 15 saniye santrifüj edildi. Altta biriken sıvı döküldü tüp tekrar filtreli tüpe takıldı ve yine aynı solüsyondan 200µL tekrar eklendi. 2 dakika maksimum hızda santrifüj edildi. Toplama tüpü sıvı ile beraber atıldı. Filtreli tüpe steril 1,5 ml’lik eppendor
tüpü takıldı. 100µL elution buffer eklendi. 10.000 rpm’de 1 dakika santrifüj yapıldı. Filtreli tüp atıldı, eppendorf tüpünde RNA hazırlandı.
t(9;22) (Light cycler t(9;22) quantification kiti)
cDNA aşaması: Kitin içinden çıkan üç standart (9a, 9b ve 9c), pozitif kontrol, negatif kontrol ve hastalar için birer 0,2 ml’lik PCR tüpleri alındı. Bu çalışmalar buz üzerinde yapıldı (+4 dereceye ulaşmak için). Her birinden tüplere 10µL alındı. Thermocyclerda 65 ˚C 10 dakika bekletildi.
Tüp sayısı kadar cDNA karışımı hazırlandı; “4,355µL su, 4µL RT buffer, 0,22µL Random primer, 0,4µL Dntp mix, 0,4µL AMV reverse transkriptaz, 0,4µL RNA az inhibitör”.
Bu karışımdan her bir 0,2’lik tüpe 10µL eklendi pipetle karıştırılırdı. Tekrar thermocycler’a götürülürdü. Aşağıdaki PCR protokolü uygulandı;
37 ˚C -60 dakika, 65 ˚C -10 dakika,
4 ˚C -10 dakika. Bu aşamadan sonra cDNA hazırdı.
LC aşaması: LC için 2 farklı karışım hazırlandı. Biri G6PDH karışımı, diğeri bcr-abl karışımı. Her bir örnek için 2 tane LC kapiller tüpü alındı. 5µL cDNA’ya 15µL G6PDH ve bcr-abl eklendi. LC cihazına yüklendi. Çalışma bittikten sonra kuantifikasyon analizi yapıldı. Analiz sonrası bcr-abl değeri olanlar pozitif, olmayanlar negatif kabul edildi. Pozitif olanlara sonuçlar verilirken bcr-abl değeri G6PDH değerine bölündü.
t (15;17) ve t(8;21):
cDNA aşaması: Transcriptör First strand cDNA synthesis kit kullanıldı. Karışım “1µL Anchored-oligo, 7µL su ile hazırlandı. 8µL karışım 0,2’lik tüplere alındı. Üzerine 5µL RNA eklendi. Thermocyclerda 65 ˚C 10 dakika bekletildi.
4 µL reaksiyon buffer, 0,5 µL inhibitör, 2 µL deoksinükleotid karışım, 0,5 µL revers transkriptase ile hazırlanan karışımdan 7µL 0,2’lik tüplere eklendi (son volüm 20µL). Tekrar thermocyclere götürüldü. Aşağıdaki PCR protokolü uygulandı;
55 ˚C-30 dakika, 85 ˚C-5 dakika,
LC aşaması: t(15;17) ve t(8;21) için ortaktır. Sadece standartlar ve primer problar farklıydı.
6,6µL su, 2,4 µL MgCl, 4 µL primer prob karışımı ve 2µL fast start ile karışım hazırlandı.
Hazırlanan karışım 15µL kapillere dağıtıldı ve üzerine 5µL cDNA eklendi. LC cihazına yüklendi.
ANALİZ: Örneklerin önce kuantifikasyon analizi yapıldı. Değer veren örneklerin melting curves analizi yapıldı. Her iki analizin uyumlu olması halinde pozitif kabul edildi. Kuantifikasyon değeri olmayanlar ve değeri olup da melting curves analizinde uygun pik vermeyenler negatif kabul edildi.
İnv (16)
cDNA aşaması: First strand cDNA Synthesis kit for RT PCR (AMV) kiti kullanıldı. Karışım 2 µL Reaction buffer, 4µL Mgcl, 2 µL Deoksinükleotid, 2µL Random primer, 1µL RNAaz inhibitör, 0,8µL ANM RT, toplam 11,8 ml 0,2 lik tüplere dağıtıldı. Üzerine 8,2µL RNA ve çalışılacaksa kontroller eklendi. Thermocyclere götürülürdü. Aşağıdaki PCR protokolü uygulandı.
25 ˚C -10 dakika, 42˚C- 60 dakika, 99˚C - 5 dakika,
4˚C -5 dakika bekletildi. Elde edilen cDNA’lar distile su ile 1/10 oranında dilüe edildi.
LC aşaması: Karışım için, 6 µL su, 2 µL Primer, 2 µL Master sybr hazırlandı. 10 µL kapillere dağıtıldı sonra 10 µL cDNA eklendi ve cihaza yüklendi.
ANALİZ: Kuantifikasyon değerlerinin melting curves analizi ile uyumlu olması gerekirdi yani değeri olan örneklerin melting curves analizine bakıldı. 90˚C de pik verenler pozitif, diğerleri negatif kabul edildi.
İstatiksel analizler, SSPS 11 bilgisayar programında yapıldı. Verilerin analizinde Mann-Whitne U testi ve chi-square testi kullanıldı. P<0,05 anlamlı olarak kabul edildi.
4-BULGULAR
Çalışmaya 41 erkek, 41 bayan toplam 82 hasta dahil edildi. Tüm hastaların yaş ortalaması 37,84±16,9 (15-76 arası); erkek hastalar için yaş ortalaması 39±17,2 ve bayan hastalar için yaş ortalaması 36,6±16,7 olarak saptandı. APL grubunda yaş ortalaması 35,00±15,9, M3 dışı lösemilerde ise yaş ortalaması 38,4±17,1 olarak saptandı. APL ve APL dışı lösemiler arasında yaş ortalaması açısından istatistiksel olarak anlamlı bir fark yoktu (p=0,47).
Hastaların 14’ü APL (9’u erkek, 5’i bayan), 68’i M3 dışı (32’si erkek, 36’sı bayan) lösemi grubundaydı
Beyaz küre sayısı ortalaması APL grubunda 3411±6450, APL dışı lösemilerde 48899±56283; nötrofil sayı ortalaması APL grubunda 2586±6368, APL dışı lösemi hastaları grubunda 10470±17578, lenfosit sayı ortalaması ise APL grubunda 554±268, APL dışı lösemi hastalarında 10539±18691 olarak saptandı. APL ve APL dışı lösemi hasta grupları arasında beyaz küre, nötrofil ve lenfosit sayıları açısından istatistiksel olarak anlamlı fark saptandı (sırası ile; p=0,003, 0,005 ve <0,0001).
Hemoglobin düzeyi ortalaması APL grubunda 8,56±1,5, APL dışı lösemilerde 8,53±2,4, hematokrit düzeyi ortalaması APL grubunda %24,73±4,9, APL dışı lösemi hastaları grubunda %24,82±7,0, trombosit sayısı ortalaması ise APL grubunda 55906±34473, APL dışı lösemi hastalarında 61884±71374 olarak saptandı. APL ve APL dışı lösemi hasta grupları arasında hemogobin, hematokrit düzeyleri ve trombosit sayıları açısından istatistiksel olarak anlamlı fark saptanmadı (sırası ile; p=0,954, 0,952 ve 0,638).
Serum LDH düzeyi ortalaması APL grubunda 410,80±292,0, APL dışı lösemilerde 854,68±702,2, serum albümin düzeyi ortalaması APL grubunda 3,12±0,6, APL dışı lösemi hastaları grubunda 3,50±0,8, APL ve APL dışı lösemi hasta grupları arasında serum LDH açısından istatistiksel olarak anlamlı fark saptanırken (p<0,0001), albümin düzeyleri açısından istatistiksel fark saptanmadı ( p=0,878).
Koagülasyon testlerininden PTZ düzeyi ortalaması APL grubunda 16,32±3,1, APL dışı lösemilerde 14,76±4,7; INR düzeyi ortalaması APL grubunda 1,36±0,3, APL dışı lösemi hastaları grubunda 1,25±0,4, aPTT düzeyi ortalaması APL grubunda
30,93±7,8, APL dışı lösemi hastalarında 27,23±4,7, Fibrinojen düzeyi ortalaması ise APL hasta grubunda 268,00±178,5, APL dışı lösemi hastalarında 274,21±118,8 olarak saptandı. APL ve APL dışı lösemi hasta grupları arasında PTZ, INR ve Fibrinojen düzeyi ortalamaları arasında anlamlı bir fark saptanmazken aPTT düzeyi ortalamasının farklı olduğu görüldü (sırası ile; p=0,134, 0,243, 0,899 ve 0,018).
14 APL hastasının 4’ünde ve 68 APL dışı lösemi hastasının 38’inde hepatomegali saptandı. Hepatomegali sıklığı açısından APL ve APL dışı lösemi hastaları arasında istatistiksel olarak anlamlı fark saptandı (p=0,04). 14 APL hastasının 6’sında ve 68 APL dışı lösemi hastasının 35’inde splenomegali saptandı. Splenomegali sıklığı açısından APL ve APL dışı lösemi hastaları arasında istatistiksel olarak anlamlı fark yoktu (p=0,570).
Toplam 82 hastanın 14’ünde, APL hastalarının tümünde t(15;17) pozitif saptandı (%17). 68 M3 dışı lösemi hastasının 5’inde t(8;21) pozitifliği (%6), 8’inde inv(16) pozitifliği saptandı (%9,7). Hiçbir hastada t(9;22) pozitifliği saptanmadı. Şekil 1- t(15;17) sıklığı Şekil 2- t(9;22) sıklığı TRAN1517 T RAN1517 1 0 P e rc ent 100 80 60 40 20 0 TRAN922 TRAN922 0 Per c e n t 120 100 80 60 40 20 0
TRAN821 TRAN821 1 0 Per c e n t 100 80 60 40 20 0 INV16 INV16 1 0 Pe rc e n t 100 80 60 40 20 0
Şekil 3- t(8;21) sıklığı Şekil 4- inv16 sıklığı
APL hastaları standart olarak ATRA-İDA (Atra 45mg/m2/gün- tedavinin 2,4,6 ve 8. günlerinde 12 mg/m2/gün İdarubisin) rejimi ile tedavi edildiler. Hastaların tümünde ortalama 25±5 günde remisyon elde edildi ve idame tedavisi (remisyon sağlandıktan sonra 3 kür antrasiklinli tedavi konsolidasyon amacıyla verildi ve idame tedavisi ATRA 45 mg/m2/gün her 3 ayda bir 15 gün, 6 merkaptopurin 90 mg/m2/gün devamlı ve metotreksat haftada bir 20 mg/m2 ) almaya başladılar. APL için remisyon kriterleri kemik iliğinde yeterli sellülaritenin ve normal maturasyonun saptanması, blast oranının % 5’in altına inmesi, Auer rod içeren hücrelerin kaybolması, çevresel kanda nötrofil sayısının 1500/mm3’den yüksek olması, trombosit sayısının 100.000 mm3’ü geçmesi ve bu tablonun en az 4 hafta devamlılığı tam remisyon olarak kabul edildi. Ortalama 12,3±9,9 aylık takip süresinde APL hastalarının tümünde remisyonun devam ettiği görüldü. Hastalarda ortalama 6,1±2,4 ayda t(15;17) negatifleşti.
M3 dışı lösemi hastalarında remisyon indüksiyon tedavisi olarak standart sitozin-arabinozid 100mg/m2x7 gün, idarubisin 12 mg/m2x3 gün tedavisi verildi. Hastaların 24’ü remisyon sonrası postremisyon tedavilerini aldı. M3 dışı lösemiler için remisyon kriterleri nötrofil sayısının 1000/mm3, trombosit sayısının 100.000 üzerine çıkması, kemik iliğinde blast sayısının % 5’in altına inmesi olarak kabul edildi. 44 hasta ise ortalama 2,4±0,8 ay sonra relaps oldular. Relaps AML hastalarında ikinci basamak tedavi olarak Flag-ida (Fludarabin 30 mg/m2 5 gün, sitozin arabinosid 2000 mg/m2 5 gün, idarubicin 8 mg/m2 3 gün) rejimi verildi. 44
hastanın 13’ünde remisyon elde edilmesine karşın 31 hasta refrakter kaldı. Refrakter olan 28’i, relaps olan 13 hastanın 8’i ve remisyonda izlenmekte iken 4 hasta hastalık veya tedavi ilişkili komplikasyonlar nedeni ile exitus oldu.
M3 dışı lösemi hasta grubunda; t(8;21) pozitif hasta grubunun tümünde tam remisyon elde edildi. t(8;21) pozitif hasta grubu ortalama 8,8±7 ay takip edilirken t(8;21) negatif hasta grubu 11,34±9,87 ay takip edildi. t(8;21) pozitif ve negatif hasta grubları arasında toplam takip sürelerine ilişkin istatistiksel bir fark oluşmadı p=(0.645). t(8;21) negatif olan 63 hastanın 43’ünde (%68,3) relaps ortaya çıktı ve 2. basamak tedavi ihtiyacı oldu. Buna karşın t(8;21) pozitif olan 5 hastanın 1’inde (%20) relaps görüldü. Relaps gelişim sıklığı bakımında t(8;21) pozitif hasta grubu negatif olan hasta grubundan daha az orandaydı ve bu oran istatistiksel fark ortaya çıkarttı p=(0,049). t(8;21) negatif olan 63 hastanın 30’unda tedaviye refrakterlik gelişirken (%47,6), t(8;21) pozitif 5 hastanın 1’inde tedaviye refrakterlik gelişti (%20). t(8;21) pozitif hasta grubunda negatif olan hasta grubuna kıyasla daha az tedaviye refrakterlik meydana gelmesine rağmen bu fark istatistiksel olarak anlamlı değildi p=(0.366). t(8;21) pozitif hasta grubunda tüm sağ kalım oranı %80 (4 hasta) iken, negatif grubta tüm sağ kalım oranı %38,1 (24 hasta) olarak saptandı. t(8;21) pozitif ve negatif hasta grupları arasında tüm sağ kalım oranları bakımından istatistiksel fark saptanmadı p=(0,08). t(8;21) negatif olan 63 hastanın 39’u (%61,9) exitus olurken ve t(8;21) pozitif olan 5 hastanın 1’i (%20) exitus oldu. t(8;21) pozitif ve negatif olan hastalar arasında exitus açısından istatistiksel fark saptanmadı p=0,151. t(8;21) pozitif olup exitus olan 1 hasta 4 ay yaşarken, t(8;21) negatif olup exitus olan 39 hasta 8,82±7,72 ay yaşamıştı. t(8;21) negatif hasta grubu daha uzun süre yaşamasına rağmen bu oran istatistiksel olarak anlamlı değildi p=(0.541).
M3 dışı lösemi hasta grubunda; inv(16) pozitif hasta grubu ortalama 9,25±7,32, inv(16) negatif hasta grubu ortalama 11,5±10,06 ay takip edildi. inv16 pozitif ve negatif hasta grubları arasında toplam takip sürelerine ilişkin istatistiksel bir fark oluşmadı p=(0.544). inv16 pozitif hasta grubunun tümünde tam remisyon elde edildi. inv(16) negatif olan 60 hastanın 40’nında relaps görüldü (%66,7) ve ikinci basamak tedavi ihtiyacı oldu. inv(16) pozitif 8 hastanın 4’ünde relaps görüldü (%50) ve ikinci basamak tedaviye ihtiyaç duyuldu. İnv(16) pozitif hasta grubunda daha az relaps görülmesine rağmen bu fark inv16 negatif hasta grubundakilerle
istatistiksel bir fark oluşturmadı p=(0.439). inv(16) negatif olan 60 hastanın 29’u tedaviye refrakter olurken (%48,3), inv(16) pozitif 8 hastanın 2’si tedaviye refrakter kabul edildi (%25). İnv(16) pozitif hasta grubunda inv(16) negatif hasta grubuna kıyasla daha az refrakterlik gelişmesine rağmen bu fark istatistiksel olarak anlamlı değildi p=(0.275). inv(16) pozitif grupta toplam sağ kalım oranı %50 (4 hasta) iken, negatif grupta toplam sağ kalım oranı %40 (24 hasta) olarak saptandı. inv16 pozitif ve negatif grupta toplam sağ kalım oranı bakımından istatistiksel fark saptanmadı p=(0,592). İnv(16) negatif hasta grubunda 60 hastanın 36’sı exitus olurken (%60), inv(16) pozitif olan 8 hastanın 4’ü exitus oldu (%50). İnv16 pozitif hasta grubunda exitus oranı inv16 negatif hasta grubundan daha az olmasına rağmen bu fark istatistiksel olarak anlamlı değildi p=(0.708). inv16 pozitif olup exitus olan 4 hasta ortalama 10,25±10,40 ay yaşarken, inv16 negatif olup exitus olan 36 hasta ortalama 8,53±7,47 ay yaşamıştır. İnv16 pozitif hasta grubu, negatif olan hasta grubuna kıyasla exitus olan hastalar daha uzun süre yaşamalarına rağmen bu fark istatistiksel olarak anlam teşkil etmemektedir p=(0.874).
t(8;21) ve inv(16) pozitif olan 13 hastada relaps gelişim oranı %38,5 iken (5 hasta), t(8;21) ve inv(16) negatif 55 hastada relaps gelişim oranı %70,9 (39 hasta) olarak saptandı. t(8;21) ve inv(16) pozitif (prognozu olumlu etkileyen translokasyonlar) hasta grubunda relaps gelişimi daha düşüktü ve bu oran istatistiksel olarak anlamlı ydı p<0,05. t(8,21) ve inv(16) pozitif olan 13 hastada tedaviye refrakterlik oranı %23,1 iken, negatif olan hasta grubunda tedaviye refrakterlik oranı %50,9 olarak saptandı. t(8;21) ve inv(16) pozitif hasta grubunda tedaviye refrakterlik daha düşük olmasına rağmen bu oran istatistiksel olarak anlamlı değildi p=(0.120). t(8;21) ve inv(16) pozitif hasta grubu ortalama 9,08±7,017 ay takip edilirken, t(8;21) ve inv16 negatif hasta grubu ortalama 11,75±10,289 ay takip edildi. Translokasyonları pozitif ve negatif olan hasta grupları arasında takip süresi bakımından istatistiksel fark oluşmadı p=(0,579). Olumlu translokasyon taşıyan grupta toplam sağ kalım oranı %61,5 (8 hasta) iken, negatif grupta %36,4 (20 hasta) idi. Toplam sağ kalım oranı bakımından her iki grupta istatiksel fark saptanmadı p=(0,09). Olumlu translokasyonların olduğu hasta grubun da 5 hasta exitus olurken %38,5, negatif grupta 35 hasta exitus oldu %63. Exitus bakımından her iki grup arasında istatistiksel fark saptanmadı p=(0,123).
5-TARTIŞMA
Akut lösemiler, lösemik hücrelerin farklılaşma ve olgunlaşma kusuru göstermeleri, normal kan hücrelerinin yapılamaması ve aşırı çoğalma kabiliyetinde olan lösemik hücrelerin, önce kemik iliğini ve ardından periferik kanı ve diğer dokuları istila etmesiyle ile karakterizedir (1). Akut miyeloid lösemi (AML); hematopoetik öncül hücrelerin (blast) heterojen klonal hastalığıdır (2). Akut lösemide sitogenetik ve moleküler genetik anomalilerin bariz bir hale gelmesi ve bunların prognostik önem göstermelerinin belirlenmesiyle yeni bir sınıflandırma ihtiyaçı olduğu gözlenmiş ve 2001 yılında Dünya Sağlık Örgütü (World Health Organizatıon, WHO) tarafından akut lösemilerde dahil olmak üzere hemopoietik ve lenfoid neoplazmaları içeren yeni bir sınıflama yapılmıştır. Akut lösemi WHO sınıflamasında; morfoloji, immünfenotipleme, sitogenetik ve moleküler biyolojik özellikler göz önüne alınmış ve akut lösemi tanısı için kemik iliğinde blastik hücre sayısı % 30’dan % 20’ye indirilmiş ve aynı zamanda nadir lösemi tipleride dahil edilmiştir (6). AML’li olgularda 100’den fazla genetik bozukluklar tanımlanmış olmakla birlikte, sıklıkla saptanan kromozomal bozukluklar t(8;21), t(15;17), inv(16), t(9;11 ) ve t(16;16)’dır. Bu kromozomal bozukluklar ve bunun varyantları yaklaşık olarak AML olgularının %40’ını oluştururlar. t(8;21) ve inv(16) translokasyonları AML’li olguların yaklaşık olarak sırasıyla %2-12 ve %1-5 kadarında görülür ve her ikisi de iyi prognozla birliktelik gösterir. Bu nedenle hastalık tanısı ve takibinde kullanılan markırlardır (65). t(15;17) AML hastalarının %5-10’unda görülür (78). Sitogenetik alt gruplar prognozu diğer herhangi bir faktörden daha iyi tanımlarlar (93). t(8;21), inv(16), t(15;17) olumlu olarak sayılmıştır.
Byrd ve arkadaşlarının yaptığı 1213 akut miyeloid lösemili hastada sitogenetik anormalliklerin tam remisyondaki prognostik etkisi, 5 yıllık relapsın oranı ve 5 yıllık tüm sağ kalım analiz edilmiş olup, tüm hastalar ortalama 8,3 yıl takip edilmiştir, inv(16) pozitifliği 96 hastada saptanmıştır (%7,9), t(8;21) pozitifliği 81 hastada saptanmıştır (%6,8), inv(16) pozitif hasta grubunda tam remisyon oranı %85, t(8;21) pozitif hasta grubunda tam remisyon oranı %91 olarak saptanmıştır, inv(16) pozitif hasta grubunda 5 yıllık relaps oranı %55 olarak bulunmuş ve bu oran normal karyotipli hastalarla karşılaştırıldığında istatiksel olarak anlamlı