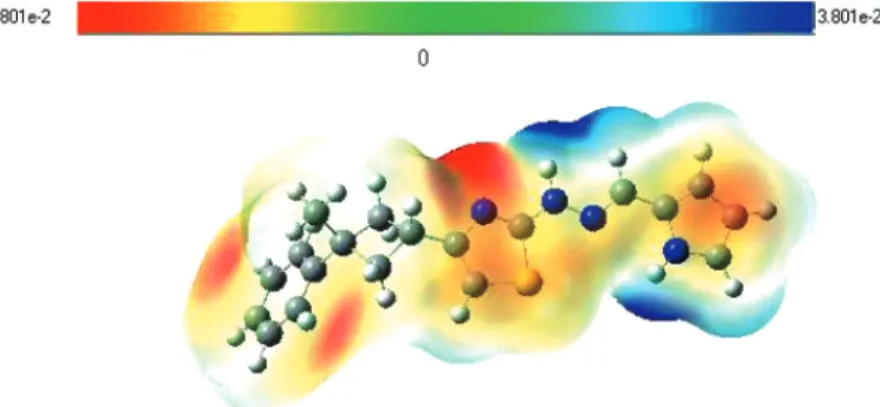
Molecular and Crystal Structure, Spectroscopic Properties of N-[4-(3- Methyl-3-Phenyl-Cyclobutyl)-Thiazol-2-yl]-N'-(1H-Pyrrol-2-Ylmethylene)- Hydrazine by Experimental Method and Quantum Chemical Calculation
Tam metin
Şekil

Benzer Belgeler
Çalışmamızda 15-15-15 gübresi; tomurcuk sayısını en çok etkileyen gübre olurken biyokütle artışını ise en çok Ozmokot (9 ay) gübresi etkilemiştir. iberica)‟nin
This study argues that the hybrid quality of the party created by a diluted version of the Ke- malism and social democracy will benefit Kiliçdaroğlu. CHP will not be divided into
Bu çalişma, Türkiye’de 2003-2009 yillari arasinda aylik olarak ortaya çikan ÜFE ve TÜFE bazli reel efektif döviz kuru değerleri için parametrik olmayan (nonpa- rametrik)
KSS faaliyetleri uygulayan şirketler için marka tercihlerini değiştirmeye bü- yük bir eğilim göstermeyen X kuşağındaki katılımcılar, söz konusu toplum zararına yönelik
Ne var ki, 15-16 Haziran işçi eylemlerinde can kayıplarının yanı sıra özellikle İstanbul’da asayişin önemli ölçüde bozulması üzerine toplanan Bakanlar
“Orhan Pamuk’u okumaya sondan başladım: Kara Kitap, Beyaz Kale, Sessiz Ev, Cevdet Bey ve Oğulları... Kara Kitap vesilesiyle diğerleri yeniden okunmuş
vefat eden Mukābele-i Süvari kâtiblerinden merhum Hattat Afif İbrahim Efendi ibn-i Mustafa Efendi’nin verâseti zevce-i menkûha-i metrûkesi Ayşe Hanım ib- netü Mustafa Bey
Kişileri hayatın getirdiği her türlü olumsuzluklara karşı koruması ve bundan daha önem lisi iç h u z u r ve asayişin sağlanarak, cem iyet hayatını ahenkli
