T.C.
DİCLE ÜNİVERSİTESİ
TIP FAKÜLTESİ
İÇ HASTALIKLARI ANABİLİM DALI
ERİŞKİN AKUT LENFOBLASTİK
LÖSEMİDE GENETİK RİSK FAKTÖRLERİNİN KANTİTATİF PCR YÖNTEMİYLE ANALİZİ VE HASTA ÖZELLİKLERİ
UZMANLIK TEZİ
Dr. Gülten ORUÇ
TEZ DANIŞMANI
Prof. Dr. M.Orhan AYYILDIZ
TEŞEKKÜR
Asistanlık eğitimimiz boyunca bilgi ve tecrübelerinden faydalandığımız, bize hekimliği öğreten çok değerli hocamız Prof. Dr. Ekrem MÜFTÜOĞLU başta olmak üzere, İç Hastalıkları ABD Başkanımız Prof. Dr.M.Emin YILMAZ’a ve bugünlere gelmemizde emeği olan diğer değerli hocalarımız; Prof. Dr. Vedat GÖRAL, Prof. Dr. Abdurrahman IŞIKDOĞAN, Prof. Dr. Kendal YALÇIN, Doç. Dr. Muhsin KAYA, Doç. Dr.Alpaslan TUZCU, Doç. Dr. Ali Kemal KADİROĞLU, Yrd. Doç.Dr.Hasan KAYABAŞI ve Yrd.Doç.Dr. M. Ali KAPLAN’ a teşekkürlerimi sunuyorum.
Tezimin oluşmasında en büyük payı olan tez danışmanı sayın hocam Prof. Dr. M. Orhan AYYILDIZ’a teşekkürü bir borç bilirim.
Çalışmamızın istatiksel değerlendirmesinde yardımlarını esirgemeyen Prof.Dr.Yusuf ÇELİK’e, DÜTF Hematoloji Laboratuarı çalışanlarından sevgili Murat YURT ve Sabahattin ASLAN başta olmak üzere tüm ekibe, hasta takibi aşamasında yardımlarını esirgemeyen sevgili asistan arkadaşlarıma, özellikle Dr. Devran IŞIK’ a ve tezimin hazırlanmasının her aşamasında her zaman en büyük desteğim ve yardımcım olan sevgili eşim Nihat’ a teşekkürlerimi bir borç bilirim.
Sevgilerimle…
Dr. Gülten ORUÇ
İÇİNDEKİLER Sayfa TEŞEKKÜR 1 İÇİNDEKİLER 2 KISALTMALAR 3 TABLOLAR 4 ÖZET 5 SUMMARY 6 1- GİRİŞ 7 2- GENEL BİLGİLER 8 2.1. Lösemi 8 2.1.1. Löseminin Tarihçesi 8 2.1.2. Löseminin Epidemiyolojisi 8 2.1.3. Löseminin Patogenezi 9 2.1.4. Löseminin Etyolojisi 9 2.1.5. Lösemilerin Sınıflandırılması 11
2.1.6. Lösemilerde Genetik İnceleme 11
2.2. Akut Lenfoblastik Lösemi 15
2.2.1. Morfolojik Sınıflama 16 2.2.2. Sitokimyasal Boyama 17 2.2.3. İmmünofenotiplendirme 17 2.2.4. Tanı 18 2.2.5. Tedavi 19 2.2.6. Prognoz 21 2.2.7. Sitogenetik 21 3- MATERYAL VE METOD 24 4- BULGULAR 25 5- TARTIŞMA VE SONUÇ 30 6- KAYNAKLAR 32 2
KISALTMALAR AL : Akut Lösemiler
ALL : Akut Lenfoblastik Lösemi AML : Akut Myeloblastik Lösemi KML : Kronik Myelositik Lösemi KLL : Kronik Lenfositik Lösemi PCR : Polimeraz Change Reaction MSS : Merkezi Sinir Sistemi SCE : Sister Kromatid Exchange FAB : French-American-British
CALLA : Common Acute Leukemia Leucosit Antigen RT-PCR : Reverse Transcriptase- Polimeraz Change Reaction HSM : Hepatosplenomegali LDH : Laktat Dehidrogenaz CD : Cluster of Differanciation Ph : Philadelphia t : Translokasyon CRP : C- Reaktif Protein UV : Ultraviole AT : Ataksi-Telenjiektazi KP : Kseroderma Pigmentozum FA : Fanconi Anemisi BS : Bloom Sendromu
KİT : Kemik İliği Transplantasyonu
KT : Kemoterapi
mRNA : Messanger (mesajcı) RNA
cDNA : Complementer(tamamlayıcı) DNA KİA : Kemik iliği aspirasyonu
WHO : World Health Organisation(Dünya Sağlık Örgütü) Ca : Karsinom
HTLV : İnsan T Hücreli Lösemi Virüsü EBV : Epstein-Barr Virüsü
TABLOLAR
Sayfa
Tablo 1: ALL’de Sayısal ve Yapısal Anomaliler ve Prognostik Önemleri.. 14
Tablo 2: FAB Sınıflaması………... 16
Tablo 3: ALL’de sitokimya……….. 17
Tablo 4: Sitogenetik anormallikler………... 23
Tablo 5: t(9;22) ile cinsiyet ilişkisi………... 26
Tablo 6: t(9;22) İle meslek ilişkisi………... 26
Tablo 7: t(9;22) ile tanı anındaki lökosit sayısı……… 26
Tablo 8: t(9;22) ile anemi ilişkisi………. 26
Tablo 9: t(9;22) ile trombositopeni ilişkisi………... 27
Tablo 10: t(9;22) ile CRP ilişkisi………... 27
Tablo 11: t(9,22) ile Sedimentasyon ilişkisi……….. 27
Tablo 12: t(9;22) ile ekstrameduller tutulum arasındaki ilişki………... 28
Tablo 13: t(9;22) ile HSM arasındaki ilişki………... 28
Tablo 14: t(9,22) ile aile öyküsü ilişkisi……… 28
Tablo 15: t(9;22) ile immünfenotip ilişkisi……… 28
Tablo 16: t(9,22) ile LDH ilişkisi……… 29
Tablo 17: t(9;22) ile morfolojik tip ilişkisi……… 29
ÖZET
Erişkin Akut Lenfoblastik Lösemi’de genetik risk faktörlerinin Kantitatif PCR yöntemiyle analizi ve bu hastaların klinik özelliklerini tanımlamak amacıyla, Ocak 2010 – Ağustos 2010 tarihleri arasında,Dicle Üniversitesi Tıp Fakültesi Hematoloji Bilim Dalı’nda Akut Lenfoblastik Lösemi tanısı konan veya daha önce tanı konmuş olup kontrol/tedavi amacıyla başvuran 39 hasta çalışmaya alındı.
Tüm hastalara moleküler hematoloji laboratuarında kantitatif PCR yöntemiyle t(9;22), t(4;11),t(12;21) ve t(1;19) bakıldı. Ayrıca immünofenotip ve klinik özellikleri araştırıldı. Daha önce tanı almış olan hastaların ise dosyalarına ulaşılarak inceleme yapıldı.Hastalardan 4’ünde t(9;22) pozitif bulundu (%10,2). Diğer 3 translokasyon ise tüm hastalarda negatif idi. 39 hastanın 35’inde immünfenotip sonuçlarına ulaşılabilindi. Bunlardan 25’i T-hücre fenotipi (%71), 8’i B-T-hücre fenotipine sahipti (%22), 2 hasta ise bifenotipik olarak değerlendirildi (%0,5). t(9;22) pozitif bulunan 4 hastadan 3’ü kadın,1’i erkekti. Bunlardan 2’si T-hücreli, 2’si B-hücreliydi. Tanı anındaki lökosit sayısı 2 hastada normal, 2 hastada ise yüksek bulundu. Hastaların 3’ünde anemi ve trombositopeni mevcuttu (%75). Hepatosplenomegali ise hastaların yarısında vardı. Ayrıca bu 4 hastanın 2’sinde ekstrameduller tutulum gözlendi. Birinde kranial, öteki hastada mediastinal tutulum vardı.Bu 4 hastadan biri,56 yaşında bayan hasta,indüksiyon tedavisi altındayken sepsisten eksitus oldu. Diğer 3 hastadan 2’si remisyonda ve idame tedavisi altındadır. Sonuncusu ise yeni hasta olup tedavisi devam etmektedir.
Erişkin ALL'de çalışılan translokasyonlar kötü risk faktörü olup özellikle t(9;22) pozitif olgularda tedaviye İmatinib eklenmesi ve hemen AlloKİT planlanması gibi özellikleri nedeniyle bu moleküler markırların tanı esnasında çalışılması oldukça önem arz etmektedir. Bizim için ilginç olan özellikle T fenotip'in literatürün aksine bizim olgularda daha fazla olmasıydı. Bunun dışında genetik anormalliklerle ilgili bulduğumuz sonuçlar literatürde ifade edilen genetik anormallik sıklığı ile paralel olarak bulunmuştur.
Anahtar Kelimeler:Akut lösemi,sitogenetik,kromozomal anomali
SUMMARY
The aim of this study is analysis of genetic risk factors in adult acute lymphoblastic leukemia with quantitative polymerase chain reaction (PCR) and to describe the clinical features of these patients; 39 patients who have an acute lymphoblastic leukemia, who were diagnosed before and applied to the Dicle University Faculty of Medicine Hematology Department, have been included in the study.
All of the patients have been tested with quantitative PCR method t(9;22), t(4;11), t(12;31), and t(1;19) in molecular hematology laboratory. Immunophenotype and clinical features have been described. For the patients who have been diagnosed before, the examination has been done through the access to their files. In 4 of the patients, t(9,22) has been found to be positive (10.2%). The other 3 translocations were negative in all patients. In 35 of 39 patients, immunphenotype results have been reached. 25 of these had T-cell phenotype (71%) and 2 had B-cell phenotype (22%), and 2 of the patients are considered as biphenotypic (0,5%). 3 of 4 patients which had t(9,22) positive were women and the one was man. 2 of these had T-cell and other 2 had B-cell. The leukocyte count during diagnosis was normal in 2 patients and high in 2 patients. Three of the patients had anemia and thrombocytopenia (75%). The half of the patients had hepatosplenomegalia. In addition, extra medullar involvement has been observed in 2 of these 4 patients. One of them had cranial the other had mediastinal involvement. One of these 4 patients, woman patient aged 56, died because of sepsis while she had induction treatment. 2 of the other 3 patients are in remission and under the maintenance treatment. The last one is a new patient and her treatment is going on.
As the translocations worked on adult ALL are bad risk factor and Imatinib is included in the treatment of t(9,22) positive cases and because of features such as immediate AlloKIT planning, it is extremely important that these molecular markers should be studied during the diagnosis. Interestingly, contrary to the literature, especially T phenotype were more in our cases. Except that, the results we have found about genetic abnormalities are found to be parallel with the frequency of genetic abnormalities which are stated in the literature.
1.GİRİŞ
Son yıllarda yapılan moleküler ve sitogenetik çalışmalarla malign hastalıkların etyopatogenezinde genetik faktörlerin çok önemli rolü olduğu açıkça görülmüştür. Bu hastalıkların tanı, tedavi seçimi ve takiplerinde bunlardan faydalanılmaktadır. Ailesel lösemi vakalarının görülmesi, bazı genetik sendromlarda lösemi sıklığının yüksek olması ve bazı lösemilerde patognomonik kromozomal anormalliğin bulunması genetik materyalin etyolojik rolüne işaret etmiştir.
Akut lösemiler, kemik iliği kan hücrelerinin hematopoezin spesifik bir evresinde matürasyonunun duraklaması ve anormal bir şekilde klonal ekspansiyonu ile ortaya çıkan malign hastalıklardır. Bu anormal çoğalan hücreler farklılaşamayıp immatür, atipik hücreler halinde kemik iliği, karaciğer, dalak, merkezi sinir sistemi ve diğer iç organlara dağılabilir.Bu arada normal fonksiyon gören hücre sayısı giderek azalır ve sitopenik durumlar ortaya çıkar. Lösemilerdeki kromozomal değişikliklerin tespiti, subtiplerin tespiti tedavi yöntemlerinin belirlenmesine yardımcı olmuştur. Ayrıca hastanın tedaviye yanıtının değerlendirilmesinde de yine sitogenetik yöntemlerden faydalanılmaktadır.Bu çalışmalarla kromozomlardaki sayısal veya yapısal mutasyonlar tespit edilebilmektedir.Bizim çalışmamızda kullanilan PCR (Polimeraz Change Reaction) yöntemi, moleküler tanı merkezlerinde rutin kullanılan sitogenetik metodlardan biridir.
Çalışmamızda, Dicle Üniversitesi Tıp Fakültesi Hematoloji Bölümü’ne belli bir süre aralığında başvuran, eski/yeni tanılı erişkin Akut Lenfoblastik Lösemi (ALL) hastalarında önemli kromozomal translokasyonların görülme sıklığını belirlemek amaçlandı.
2. GENEL BİLGİLER 2.1.LÖSEMİ
2.1.1. Löseminin Tarihçesi
1827’de Velpeau tarafından lösemi ile ilgili ilk vaka bildiriminden bu yana, lösemi gerek klinik gerekse genetik incelemelerde ilgi duyulan bir neoplazm olmuştur. Velpeau tarafından tespit edilen hastada; ateş, güçsüzlük, karın şişliği ve idrar yolları taşlarından kaynaklanan ağrılar şikayet edilmiş ve hastaneye başvurusundan kısa bir süre sonra hasta kaybedilmiştir. Hastanın otopsisinde çok büyük dalak ve karaciğerin yanı sıra, kanın kırmızı şarap renginde ve kıvamının artmış olduğu adeta lapa kıvamında olduğu bildirilmiştir (1). Bu tarihten 1845 yılına kadar lösemi çok iyi tanımlanamamıştır. Bu tarihlerde Virchow ve Benett ayrı ayrı olgularının otopsisinde, kanın beyaz rengine dikkat çekmiş ve hastalığı tanımlamışlardır (2). Bu yüksek lökosit sayısına atfen beyaz kan terimi (weisses blut) kullanılmış, daha sonralarda ise Yunanca kökenli olarak beyaz anlamına gelen “leukos” ve kan anlamına gelen “haima” sözcüklerinden leukemia terimi türetilerek kullanılmaya başlanmıştır (2).
Lösemiler kan hücrelerinin üretildiği kemik iliğinin klonal habis hastalıklarıdır. Çok fazla çoğalan fakat farklılaşmayan lökositler, başta kemik iliği olmak üzere karaciğer, lenf bezleri, dalak, deri, testis ve merkezi sinir sistemi (MSS) gibi organ ve sistemleri işgal edebilir. Yeterinden fazla hücre üretilmesine karşın, bu hücreler tam olarak olgunlaşıp farklılaşmadığından işlev gören hücre sayısı azalarak anemi, trombositopeni ve nötropeni oluşmaktadır (3). Bu tam olarak farklılaşmamış hücreler ‘blast’ olarak adlandırılmaktadır. Bu hücreler normal fonksiyonlarını yerine getirme yeteneğinden yoksundur. Lösemilerin kendi içindeki alt sınıflamalarında ve fazlara ayrımında bu blastların normal hücrelere oranı ve yapısı dikkate alınmaktadır.
2.1.2. Löseminin Epidemiyolojisi
Akut Lenfoblastik Lösemi(ALL), çocukluk çağında en sık görülen malignite olup Amerika Birleşik Devletleri’nde 15 yaş altında tanı alan kanserlerin %23’ünü oluşturduğu bildirilmektedir (4,5).Erişkinlerdeki akut lösemilerin ise %20’sini oluşturur(4,5). Gelişmiş ülkelerde akut lösemilerin % 83’ünü ALL, % 17sini AML(Akut myeloblastik lösemi) oluşturmakta olup, insidansta bölgesel farklılıklar görülebilir. Ülkemizde kanser istatistikleri henüz yeterince sağlıklı olmamakla birlikte, ALL(çocuklarda) sıklık açısından yine birinci sırada yer almakta ve çeşitli merkezlerde tedavi gören hastaların en az % 27’sini oluşturmaktadır (6). Görülme sıklığı en fazla 3-5 yaş arasındadır ve daha sonra sıklık giderek azalarak yaşamın üçüncü dekatında yeniden artar. ALL’nin erken çocukluk çağında sık görülmesinin etiyolojik önemi açık değildir. Ancak gebelikteki olaylar ve immün sistemin gelişmesi ile ilişkili olduğuna dair görüşler vardır (7). ALL erkeklerde kızlardan daha sık görülür.Erkek/Kadın oranı 1,4/1,0’dır. T hücreli ALL’de ise bu erkek predominansı daha belirgindir (4:1). Cins prognoz açısından önemli bir gösterge olup kızlardaki sürvi oranı daha yüksektir.
2.1.3. Löseminin Patogenezi
Kan hücreleri hematopoietik organlardaki kök hücrelerinden gelişirler. Fetal dönemde karaciğer ve dalak gibi geçici hematopoietik organlarda oluşturulan kan hücreleri fetal dönemin sonuna doğru esas hematopoietik doku haline gelen kemik iliğinden üretilir.
Tüm kan hücrelerinin kemik iliğinde tek bir kök hücresinden geliştiğine inanılmaktadır. Bu hücreler bütün hücre türlerini verdiği için ‘pluripotent kök hücresi’ olarak isimlendirilir. Pluripotent hücreler çoğalarak tek tür kan hücresini oluşturan ana hücreleri verirler. Bu hücrelerden bir kısmı ‘lenfoid’ diğer bir kısmı ise ‘myeloid’ hücre serisine farklılaşır. Myeloid hücreler kemik iliğinde gelişerek eritrositler, granülositler, monositler ve megakaryositlere dönüşürler. Gelişimin erken döneminde lenfoid hücreler kemik iliğinden lenf düğümleri, dalak ve timusa göç ederek lenfositlere farklılaşırlar.Yani hematopoez kök hücrelerinden gelişen ve farklılaştıkça dönüşme özellikleri azalan eş zamanlı ve devamlı olarak çeşitli hücrelerin üretilmesi ve bu hücrelerin farklılaşması işlemidir. Lösemi, kök hücreden spesifik kan hücrelerinin oluşumuna kadar herhangi bir farklılaşma aşamasında gözlenebilir. Lösemik dönüşüm temelde bir mutasyon sonucu oluşmaktadır. Mutasyona uğrayan hücrelerin çoğu organizma tarafından çeşitli mekanizmalarla ortadan kaldırılmasına rağmen az sayıda hücre aşırı çoğalma özelliği kazanabilmektedir. İşte bu hücreler “klonojenik lösemik hücreler” olarak adlandırılmaktadır (2). Lösemilerin tek hücre kaynaklı klonal bir genişleme gösterdiği düşünülmektedir.
2.1.4. Löseminin Etiyolojisi
Lösemilerin oluş sebebi tam olarak bilinmemekle birlikte meydana gelişinden tek bir faktörün sorumlu olmadığı da bir gerçektir. Muhtemelen çeşitli etkenler hastalığın oluşumunu hazırlamaktadır. Lösemi oluşumuna götüren etmenleri 2 grup altında inceleyebiliriz.
1) Çevresel (dış) etkenler 2) Bireysel (kalıtsal) etkenler 2.1.4.1. Çevresel Etkenler
Fiziksel ve kimyasal ajanlar ile virüsleri içeren çevresel etkenlerin karsinojenik etkisinin olduğu bilinmektedir (8,10). Bu karsinojenlerin kromozomlar üzerinde hasarlar yaparak kanser oluşturabildikleri bildirilmiştir (9,11). Karsinojenlerin kanser oluşturma riskleri; karsinojenlerin dozuna, etki süresine ve kişinin gen duyarlılığına bağlanmaktadır (12, 13). Örneğin Çernobil faciasında ülkemizin bölgeye en yakın yöresindeki Karadeniz kıyılarında artan sitogenetik değişim oranı buna en güzel göstergedir (14). Özellikle radyasyon, hücrede öncül nükleotidlerin yapısında değişikliğe neden olmaktadır. Bu da kromozomlarda kırılmalara, ring kromozom, disentrik kromozom ve sayısal anomalilere neden olarak lösemi insidansını arttırmaktadır (15,16).
Ayrıca DNA’da fosfodiester bağlarında kırılmalara neden olduğundan DNA sentez evresinde hatalı replikasyon ve dolayısıyla kromatid tipinde kırıklar oluşturmaktadır.Bunların yıllar içindeki artış ve birikimi artmış lösemi insidansı olarak geri bildirilmektedir. Örneğin; atom bombasının etkisine maruz
kalanlarda 3 yıl sonra lösemi görülmeye başlanmış ve görülme oranı 5-7 yıl sonra oldukça fazla artmıştır.
Ayrıca spondilit artrit için radyasyon gören vakalarda da lösemi insidansı yükselirken (11, 17), yüksek doz radyasyonun yalnız lösemi insidansı değil diğer malignitelerde de artışa sebep olduğu gözlenmiştir (8, 9, 17). Yaklaşık 3000 A0 dalga uzunluğundaki UV(ultraviyole) ışınlarının özellikle beyazlarda deri kanserine neden olduğu bildirilmiştir.
Günümüzde en çok viral etyoloji üzerinde durulmaktadır.Kanser etyolojisinde rol oynadığı düşünülen virüsler:Retrovirüsler, EBV(Endemik Burkitt, nasofaringeal Ca), HTLV-1 (Erişkin T-hücreli lösemi), HTLV-2 (Hairy cell lösemi) dir(20).
Birçok kimyasal ajan da radyasyon gibi karsinojenik etki edebilmektedir. Ayrıca kimyasal karsinojenler hücre bölünmesi sırasında kromozom davranışlarını etkilemektedir. Kimyasal karsinojenlerin en önemlileri benzen ve alkilleyici ajanlardır.Benzenin hematopoietik, lenfoid ve akciğer kanserlerine sebep olduğu vurgulanmaktadır (8, 18). İstanbul’da benzene maruz kalanlarda lösemi insidansı normalden 2-3 kat daha fazla bulunmuştur (19). Benzer olarak kloramfenikol ve fenilbutazonun lökomojenik etkisini gösteren raporlar vardır (21, 22).
Yine kombine ilaç tedavisinde, pestisit ve insektisit gibi kimyasal ajanların kullanımında, artan lösemi insidansı çalışmalarda göze çarpmaktadır (23). Kombine terapide antitümör ajanlar, kromozomal bozulma ve kemik iliği disfonksiyonuna neden olurlar, alkilleyiciler ise immünosüpresif özelliğe sahiptirler (24,25). Kromozom veya kromatidlerin yanlış birleşmelerinden dolayı anormal kromozom sayıları da meydana gelmektedir (17).
2.1.4.2. Kalıtsal Etkenler
Malign hastalıkların etiyolojisinde genetik materyalin önemli rol oynadığı moleküler ve sitogenetik gelişmelerle net bir şekilde açığa çıkmaktadır. Özellikle familyal lösemi olgularının tahmin edilenden daha sık olması genetik faktörün lösemi etiyolojisinde rol oynadığı lehinde bir delildir (26). Bu genetik eğilimi destekleyen diğer iki unsur da, bazı kalıtsal hastalıklarda lösemi sıklığının artmış oluşu ve monozigot ikizlerde bir kardeşte lösemi görüldüğü zaman diğer kardeşte de lösemi görülme insidansının yüksek oluşudur (27). Çift yumurta ikizlerinde bu oranın düşüşü genetik yapının da olayla ilişkili olduğunu desteklemektedir. Doğumsal kromozomal anomalileri bulunan olgularda malignite oranı sağlıklı bireylere göre yüksektir (15, 28). Down sendromlu olgularda lösemi insidansı, sağlıklı bireylere göre 16-30 kat daha fazladır (27, 29, 30). Çocukluk döneminde Down sendromlu olguların; % 30,2’si AML’ye, %69,8’i ALL’ye, yeni doğan dönemde ise % 80 AML’ye, % 20 ALL’ye dönüş olduğubildirilmiştir (9, 11, 28). Aynı şekilde, Klinefelter ve Turner sendromlu olgularda da eksik veya fazla kromozomların neden olduğu gerek hormonal gerekse genetik etkilerden dolayı çeşitli tip kanserlere yakalanma oranlarının sağlıklı bireylere göre katlarca fazla olduğu birçok çalışmada gösterilmiştir (9, 10,28). Otozomal resesif kalıtım gösteren Ataksi telenjiektazi (AT), Kseroderma pigmentozum (KP), Fanconi anemisi (FA), Bloom sendromu (BS) gibi durumlarda, çeşitli tip kanser vakalarında önemli artışlar gözlemlenmiştir. BS’lu hastalarda; homolog rekombinasyon eğilimi artar, quadriradial formasyon, SCE(Sister Chromatid Exchange=Kardeş
Kromatid Değişimi) sıklığında artış vardır. FA hastalarda; somatik hücrelerde kromozom aberrasyonlarında genel bir artış vardır, SCE sıklığı normaldir. AT hastalarında ise strüktürel kromozom aberrasyonlarında artış vardır, stabil düzensizlikler bulunur fakat normal SCE görülür. KP hastalarında temel anormallik DNA tamir mekanizmasındaki azalmadır. Görüldüğü gibi kromozomal yapıdaki anormallik ve kararsızlıklar bu hastaları normal bireylere oranla daha hızlı kanserojen oluşumuna sürüklemektedir (9, 11,15). Bazal şartlarda bu kromozom kırık sendromlarında kromozomal aberrasyonlar normal seviyededir, fakat sistem uygun mutajence baskı altına alınırsa o zaman anomalilerde bir artış olmaktadır.
2.1.5. Lösemilerin Sınıflandırılması
Lösemiler, köken aldıkları hücre grubuna, semptomlarına, ortaya çıkış ve ilerleme hızlarına ayrıca klinik seyirlerine göre gruplara ayrılırlar. Lösemiler ortaya çıkış hızlarına göre öncelikle akut ve kronik olmak üzere 2 ana gruba ayrılırlar. Her iki grup da kendi içinde köken aldıkları hücre grubuna göre ikişer alt sınıfa ayrılır. Böylece lösemiler 2 ana başlıkta 4 grupta incelenir.
1. Akut Myeloblastik Lösemi (AML) 2. Akut Lenfoblastik Lösemi (ALL) 3. Kronik Myelositer Lösemi (KML) 4. Kronik Lenfositer Lösemi (KLL)
Her bir lösemi tipi de kendi içinde gelişiminin herhangi basamağında ya da evresinde olduğuna göre subtiplere ayrılırlar.
2.1.6.Lösemilerde Genetik İnceleme Moleküler Genetik Teknikler: - Klasik Sitogenetik
- FISH - PCR
- Southern Blotting - Western Blotting
- mRNA’nın insitu hibridizasyonu
PCR tekniği çok küçük miktardaki DNA’nın kullanımına imkan veren ve
oldukça hassas olan bir teknik olması açısından fazlaca tercih edilmektedir. Bir DNA kaynağında bulunan hedef DNA zincirlerinin hızlı ve çok yönlü olarak çoğaltıldığı in vitro bir yöntemdir. DNA’nın amplifikasyonu sonucu hedef dizinin 105 kopyası elde edilebilmektedir. Teknik ile 104 ile 108’de 1 (1/104-108) oranında anormal hücre tespit edilebilmektedir.
Kantitatif PCR ve Reverse Transcriptase PCR (RT-PCR) gibi farklı uygulama tipleri de vardır. RT- mutasyon taramalarında PCR özellikle kullanılır ve teknikte komplementer DNA kullanılmaktadır. Çalışılacak dokudan mRNA izole edilmekte ve bu mRNA’dan reverse transkriptaz enzimleri yardımıyla cDNA’lar elde edilmektedir (48, 49). Elde edilen cDNA, bundan sonraki PCR için kalıp görevi görür. Teknik ile hızlı, hassas ve spesifik tetkikler yapılabilmektedir. Amplifikasyon miktarına göre 105-106 normal hücre içindeki 1 Ph(+) hücre tespit edilebilmektedir (50, 51).
2.1.7.Kromozomal Değişim Terminolojisi
ALL’de görülen kromozomal anomalileri genetik olarak incelemeden önce konuyu daha iyi anlayabilmemiz için gerekli terminolojiyi bilmek gereklidir. Kromozomlar sayısal ve yapısal olarak adlandırılan iki tip düzensizliğe sahip olabilmektedirler. Bunlar ayrı ayrı özel bir terminoloji ile açıklanır.
Özelliklerin kuşaktan kuşağa değişmeden aktarılmasını sağlayan kromozomlar; şekil, büyüklük ve sayı gibi birçok yönden her canlı türü ve o tür içindeki ait olduğu kişi bakımından sabit ve ayırt edici niteliktedir. Normalde 46 olan (2n=46) insan kromozomları kimi zaman hem sayı, hem şekil hem de yapı bakımından değişiklik gösterebilir. Bu kromozomal düzensizlikler gerek mikroskobik gerekse moleküler boyutta yeni teknikler ile (Bantlama, FISH, RTPCR) tespit edilebilmektedir (25). Kromozom kusurlarını ana hatları ile sınıflandırılacak olursak; kromozom sayısı mutasyonları ve kromozom yapısı mutasyonları şeklinde iki başlık altında özetleyebiliriz.
2.1.7.1.Kromozom Sayısı Mutasyonları
Normal bir mayoz sonucunda oluşan gametler 23 adet kromozom içerirler. Bu sayı insan için haploid (n) sayıdır. Vücut hücrelerindeki kromozom sayısı ise haploid sayının iki katı olup, diploid (2n=46) olarak adlandırılır. Kromozom sayısı düzensizlikleri de kendi içerisinde 2’ye ayrılır.
Kromozom sayısındaki artış yada azalışlar temel kromozom sayısının (n=23) tam katları kadar oluyorsa buna öploidi ve sayıya da öploid denir. Bu durumda hücrelerdeki kromozom sayısı, o organizma türü için normal olan haploid sayısının tam katı kadar artmıştır. Bir öploidi tipi olan haploidi eşey hücrelerindeki temel kromozom sayısını gösterir (n=23). Diğer bir tip; iki eşey hücresinin birleşmesi ile elde edilen kromozom sayısıdır ki bu hücrelere diploid hücreler denir ve insan için 2n=46 kromozom içerirler. Diğer bir tip triploidi (3n=69) olarak adlandırılır ve temel kromozom sayısı üç kat artmıştır. Triploid hücreler 69 kromozom içerirler. Tetraploidi (4n=92) durumunda ise temel kromozom sayısı dört kat artmış olup hücreler 92 kromozom içermektedirler. Kuramsal olarak temel kromozom sayısını sonsuza kadar arttırmak mümkün iken diğer ploidiler insanda gözlenmemektedir.
Bazı durumlarda ise mozaik durumlar gözlenebilmektedir. Böyle bir durumda farklı hücrelerde farklı( n) sayılarda kromozom bulunabilmektedir bu durum miksoploidi olarak adlandırılır (52). Kromozom sayısındaki artış yada azalışlar temel kromozom sayısının tam katları kadar olmayıp, bir veya birkaç kromozomun eksikliği veya fazlalığı şeklinde ise bu duruma anöploidi ve sayıya da anöploid denir. Anöploidi olguları öploidi olgularından daha sık gözlenir ve pek çok kromozomal sendromun oluşmasına sebebiyet verirler. Anöploidi, kromozomlardaki artma ya da eksilmeye göre ikiye ayrılır.Birincisi hiperploidi olup; insanlardaki diploid sayıdan bir yada daha çok sayıda fazla kromozom bulunması durumudur (Örneğin: 2n+1).
Trizomi en sık rastlanılan hiperploidi tipidir ve insan kromozomlarından herhangi birinin iki yerine üç tane bulunması durumudur yani 2n+1=47 kromozomlu bir karyotip söz konusudur. Tetrazomi daha nadir gözlenir. Tetrazomik durum ya belli bir homolog kromozom çiftinden iki yerine dört tane bulunmasıyla ya da iki ayrı homolog çiftin trizomisi ile ortaya çıkar. İkinci tip
hipoploidide; karyotipteki diploid sayıdan bir yada daha çok kromozomun eksilmesidir (Örneğin: 2n-1).
Hipoploidide kromozom çiftlerinden birinin bulunmaması (2n-2) nullizomi olarak adlandırılırken, diploid kromozomdan yalnız bir kromozomun eksik olması (2n-1) monozomi olarak adlandırılır (52).
2.1.7.2.Kromozom Yapısı Mutasyonları
Sayısal düzensizliklerin oluş nedeni hücre bölünmesindeki kusurlar iken yapısal düzensizliklerin nedeni aynı ya da değişik kromozomlardaki kırılma ve yeniden düzenlenmelerdir. Yapısal kromozomal değişikliklerin başlıcaları şunlardır:
1.Yer Değiştirme (Translokasyon)
Bir kromozomdan kopan parçanın diğer bir kromozoma yerleşmesidir. Translokasyonlar üç grup içinde incelenebilir.
• Karşılıklı translokasyon
• Sentrik kaynaşma tipi translokasyon
• İnsersiyonel translokasyon (Transpozisyon)
Karşılıklı translokasyon (resiprokal translokasyon): Bir kırılma sonucu,homolog yada homolog olmayan kromozomlardan kopan parçaların karşılıklı yer değiştirmesine denir.
Sentrik kaynaşma tipi translokasyon: Akrosentrik kromozomlarda görülen özel bir resiprokal translokasyon tipidir. Bu translokasyonda kromozomlardan sentromere yakın kısa kolunda, diğerinde ise yine sentromere yakın fakat uzun kolunda birer kırılma olur. Daha sonra iki kromozomun uzun ve kısa kolu birleşerek translokasyon kromozomlarını oluştururlar. Bu translokasyona Robertsonian tipi translokasyon da denilmektedir (53).
İnsersiyonel translokasyon (Transpozisyon): Homolog olmayan iki kromozomdan birinde iki noktada, diğerinde ise bir noktada kırılma olur. İki kırılma noktası olan kromozomdaki parça tek kırılma olan kromozoma girer ve kaynaşır. Bu kromozomlardan birinde artma diğerinde ise eksilme söz konusudur.
2. Eksilme (Delesyon)
Bir kırılma sonucu, kromozomun küçük bir parçasının kopması demektir. Kopma iki türlü olabilir; ya bir darbe sonucu kırılan kromozom parçası kopar (terminal delesyon), yada iki darbe sonucu kopan parça aradan ayrıldıktan sonra iki parça yeniden kaynaşır (interstisyel delesyon) (52).
3. Artma (Duplikasyon)
Homolog iki kromozomdan birinde çift darbe sonucu kopan parça, diğerinde tek darbe sonucu kopan aralığa girerek kaynaşacak olursa artma yada duplikasyon olgusu ortaya çıkar. Duplikasyon kendini iki tipte gösterir: Homolog kromozomlarda artmada gen duplikasyonu olur. Tandem duplikasyonda genler ard arda dizilmiştir. Ters tandem duplikasyonda artan parça tersine dönerek yine yerine yerleşmiştir (53).
4. Ters Dönme (İnversiyon)
Bir kromozoma iki darbenin gelmesi ve bunun sonucunda kopan parçanın kaybolmadan kendi ekseni çevresinde 1800 dönerek yine eski yerine yapışmasına ters dönme yada inversiyon denir. İki türlü ters dönme olmaktadır: • Parasentrik inversiyon; sentromerin dışında olan ters dönmelere verilen addır. • Perisentrik inversiyonda ise; iki darbe uzun ve kısa kollarına gelir, sentromeri içinde bulunan parça kendi çevresinde 1800 dönerek eski yerine yapışırsa perisentrik ters dönme ortaya çıkar (53).
5. Halka (Ring) Kromozom
Bir kromozomun iki ucunda iki darbe sonucu kırıklar olur ve bu kırık uçlara başka bir parça birleşmeden iki uç kaynaşırsa halka şeklinde bir kromozom ortaya çıkar ki buna halka yada ring kromozom denir (53).
6. İzokromozom
Anafazda birbirinden ayrılarak çekilecek olan kromatidler bazen boylamasına değil enlemesine bölünebilir. Bu durumda yavru hücrelerden birinde yalnızca kromozomun kısa kolları bulunurken, diğerinde yalnızca uzun kollar bulunur. Böyle sentromerin enlemesine bölünmesi sonucu ortaya çıkan median görünümlü kromozomlara izokromozom denir (52,53).
Tablo 1: ALL’de Sayısal ve Yapısal Anomaliler ve Prognostik Önemleri Hiperdiploidi 51-65 kromozom); en sık rastlanılan sayısal kromozomal
anomalidir ve iyi prognoz göstergesidir. Tetraploidi (82-94 kromozom); kötü prognoza işaret eder. Hipodiploidi (<46 kromozom); kötü prognoza işaret eder.
Psödodiploidi (yapısal anomali gösteren 46 kromozom); kötü prognoza işaret eder.
t(12;21) Pediatrik öncül B-ALL’lerin %25’i, TEL/AML1 gen değişimi, iyi prognoz, genellikle standart sitogenetik analiz ile
yakalanmaz, moleküler yöntemlerle bakılmalıdır. t(9;22) Ph
kromozomu
Pediatrik ALL’lerin %5’i, erişkin ALL’lerin %30’u, BCR/ABL gen değişimini taşır. Kötü prognoz; sıklıkla miyeloid antijen ekspresyonu ile birliktedir.
t(1;19) Pediatrik ALL’lerin %5’i; B-öncül hücre fenotipi, E2A/PBX1, daha önce kötü prognoza işaret ederken agresif tedavi sonrası prognoza katkısı anlamlılığını yitirmektedir.
t(4;11) Pediatrik ALL’lerin %4-8’i, MLL gen değişimi; birçok eşdeğeri,
infantil ALL’de sıktır (1 yaş); kötü prognoz; sıklıkla CD10, CD15+ fenotip
t(8;14), t(2;8), t(8;22)
Pediatrik ALL’lerin <% 5’i, c-MYC onkogeni; immünoglobulin, Burkitt hücreli lösemi ile ilişkilidir; agresif genleri kemoterapi öncesi dönemde kötü prognoz göstergesiydi; agresif tedavi ile daha az önemli, daha iyi sonuçlar alınır
2.2. AKUT LENFOBLASTİK LÖSEMİ
Akut lösemiler (AL), hematopoietik dokuların malign, ilerleyici ve tedavi edilmedikleri takdirde genellikle 6 ay içinde ölümle sonuçlanan bir grup hastalığıdır. Hastalık sitopatolojik olarak kemik iliği ve çevre kanında iri, olgunlaşmamış ve anormal hücrelerin bol miktarda bulunması ile karakterizedir. Bu anormal hücreler kemik iliğini infiltre ederek diğer hücrelerin yerini alır, kemik iliğinin normal yapısını tamamen bozabilir. Hastaların %10’unda lökosit sayısı normal iken, %80-90’ında periferik yaymada blastlar görülür (9, 10). Blast oranı prognozu belirleyen en önemli faktördür. Geliştikleri hücre grubuna göre myeloid veya lenfoid olarak 2 alt gruba ayrılırlar. Bazı hastalarda lösemik transformasyon myeloid kök hücre düzeyinde bazı hastalarda ise yönlendirilmiş hücrelerde meydana gelmektedir (16, 31, 32).
AL’ler prognostik faktörlere ve morfolojisine göre ALL ve Akut nonlenfoblastik lösemi (ANLL)=AML olmak üzere 2 gruba ayrılır. AL’ler her yaşta görülebilen hastalık olup, çocuklarda 1-14 yaşlar arası ölümcüldür ve kendini ALL tipinde gösterirken, erişkinlerde ise daha sık olarak AML tipinde görülmektedir. Yaş ilerledikçe AML sıklığı artarken ALL sıklığı azalır. Beyaz küre sayısı olguların % 80-90’ında artmış olup 20.000/mm3’ den fazladır. Olguların büyük bir kısmında anemi ve trombositopeni vardır.
Bu ana gruplarda da bazı alt gruplar vardır. Bu alt gruplar muhtemelen hematopoietik öncülerin hangi aşamada lösemik hale geldiklerini yansıtmaktadır. Ayrıca sınıflandırmada, hematopoietik sistemde anormal prolifere olan hücrelerin morfolojik, immünolojik ve sitokimyasal özellikleri dikkate alınmalıdır.
Burada kullanılan yararlı bir sınıflandırma sistemi French-American-British (FAB) çalışma grubunun yaptığı sınıflandırmadır. Sınıflandırma esas olarak rutin boyalarla boyanan preparatların morfolojik incelenmesine ve sitoşimik reaksiyonlara dayanmaktadır (9, 10, 15).
ALL, lenfoid öncü hücrelerin klonal ekspansiyonu sonucu gelişir. Malign transformasyon farklılaşmanın değişik safhalarında gelişebilir. Akut myeloblastik lösemi blastları ile karşılaştırıldığında lösemik ALL blastlarının farklılaşma ve olgunlaşma kapasitesi oldukça sınırlıdır. Klonalite, kromozom analizleri, yüzey proteinleri analizi, immunoglobulinler ve Thücre reseptörlerini kodlayan genlerin yeniden düzenlenme analizleri ile gösterilebilir. Kemik iliğinde blast sayısı arttıkça hematopoez geniş ölçüde baskılanır. Anemi, lökopeni, trombositopeni ve buna bağlı olarak da solukluk, enfeksiyonlar, kanama ve kemik ağrıları ile başlar. Lösemik hücrelerle infiltrasyon sonucu birçok hastada lenfadenopati ve hepatosplenomegali görülür. Testiküler infiltrasyon ve MSS’nin lösemik infiltrasyonu hastaların % 10’unda görülür. Periferik kan tetkiklerinde vakaların % 50’sinde 10x109/L üzerinde lökositoz ve %80 vakada trombositopeni (100x109/L altında) görülür. Lösemik blastlar
periferikkan yaymasında görülmeyebilir, kemik iliği aspirasyonu gereklidir. ALL’de lenfoblastlar dar, sitoplazma agranülerdir (33).
2.2.1.Morfolojik Sınıflama
FAB sınıflamasına göre lösemik hücrelerin L1, L2 ve L3 olmak üzere 3 subtipi vardır. Bu sınıflamada daha çok sitolojik özellikler baz alınmıştır.
L1 alt tipi daha çok çocuklarda görülmekte; monomorfik, yuvarlak çekirdekli, dar sitoplazmalı, küçük ve orta büyüklükte blastlar, homojen nükleer kromatin yapısı ve belirsiz nükleoluslar dikkat çekicidir.
L2 alt tipine erişkinlerde sıklıkla rastlanır. Blastlar daha büyük, daha geniş sitoplazmalı ve değişken görünümlüdür. Düzensiz kenarlı çekirdek yapısı ve belirgin nükleuslar görülmektedir.
L3 tip çok nadir görülür, hücreler koyu bazofilik sitoplazma ve sitoplazmik yağ içeren vakuoller ile karakterizedir.
Morfolojik sınıflama giderek önemini kaybetmektedir. Bunlar yerine, monoklonal antikorların kullanıldığı yüzey belirleyici analizi, sitoplazmik belirleyiciler, kromozom yapısı ve yeniden gen düzenlenmelerinin analizine başvurulmaktadır. Bu çalışmalar ALL’nin sürekli olarak tanımlanan yeni alt grupları ile büyük orandaki heterojenitesini ortaya koymuştur. Özellikle ALL’nin AML’den ayırımında ve kendi içinde alt tiplere ayırımında monoklonal antikorlardan yararlanılır (34).
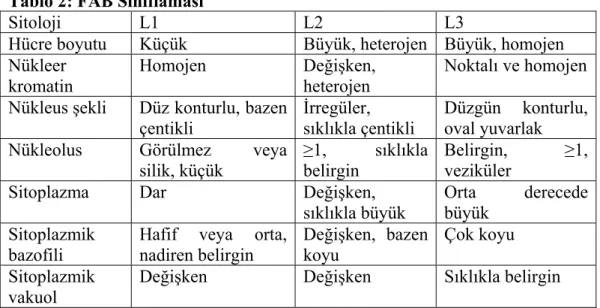
Kromozomal çalışmalarda da prognostik önemi olan yapısal ve sayısal değişiklikler tanımlanabilmektedir. Ayrıca özgül yapısal sitogenetik çalışmalar normal hücresel fonksiyonlar ve onkogenezde rol oynayan genlerin belirlenmesini sağlamıştır (35). Yeni WHO sınıflaması ALL subtiplerini fenotipik ve sitogenetik özelliklerine göre sınıflamaktadır. Yani FAB sınıflaması bu sınıflamada kullanılmaktadır (Tablo-2).
Tablo 2: FAB Sınıflaması
Sitoloji L1 L2 L3
Hücre boyutu Küçük Büyük, heterojen Büyük, homojen Nükleer
kromatin
Homojen Değişken, heterojen
Noktalı ve homojen Nükleus şekli Düz konturlu, bazen
çentikli
İrregüler, sıklıkla çentikli
Düzgün konturlu, oval yuvarlak
Nükleolus Görülmez veya silik, küçük
≥1, sıklıkla belirgin
Belirgin, ≥1, veziküler
Sitoplazma Dar Değişken, sıklıkla büyük
Orta derecede büyük
Sitoplazmik
bazofili Hafif veya orta, nadiren belirgin Değişken, bazen koyu Çok koyu Sitoplazmik
vakuol Değişken Değişken Sıklıkla belirgin L1 tip ALL: Küçük ve homojen hücrelerden oluşur. Hücrelerin %25’i T lenfosittir. Nükleus membranı düzenlidir. En sık görülen kromozom anomalileri t(9;22), t(1;19), t(4;11), t(8;14), 6q ve 9p anormallikleridir.
L2 tip ALL: Hücre sitoplazması daha geniş olup, bir veya daha fazla belirgin nükleolus vardır. Hücrelerin %25’i T lenfosittir. Karşılaşılan kromozom anomalikleri t(9;22), t(4;11), 6q, +21, +8, i(17q), 7p-,11q-‘dir (36, 37).
L3 tip ALL: Hücreler büyük ve homojendir. ALL’nin %5’inden fazlası bu tiptir. Pek çok hücre olgun B lenfositlerden köken alır (11, 19, 21, 38,39). Kromozom anomalileri t(8;14), t(8;22), t(2;8), 1q-, +6, 6q-, 8q+ dir.
ALL 5 yaşın altındaki çocuklarda daha sık görülür (36, 41). Erişkin ALL’de ise remisyon oranı ve yaşam süresi çocuklara göre daha azdır. t(9;22) (q34;q11) ALL’de sık gözlenen bir kromozomal düzenlemedir. Yaklaşık olarak yetişkin ALL’ lerin %20’sinde, çocuk ALL’lerinin de %5’inde görülür. Sitogenetik düzeyde kopma noktaları KML’ ye benzemektedir (42). ALL vakalarında BCR (Breakpoint Cluster Region)’nin kırılma noktası Major BCR olarak adlandırılan bölgeye yerleşmektedir. Buna uyan protein p210 proteinidir veya minör BCR olarak adlandırılan bölgeye yerleşmektedir ki buna uyan protein de p190 proteinidir. Oysa KML vakalarının büyük çoğunluğunda Major BCR bölgesinde kırılma olmakta ve ilgili protein p210 olmaktadır (43,44).
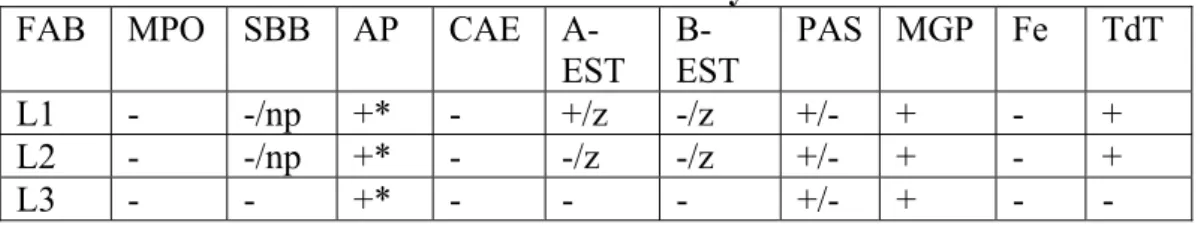
2.2.2.Sitokimyasal Boyama
Blastların bazı boyaları alıp almama veya az ya da çok almaları da lösemi tipini belirlemek için faydalı bir özelliktir. Miyeloperoksidaz (MPE), Sudan siyahı (SBB), Periyodik-Aside-Schiff (PAS), Asid Fosfataz (AP), Nonspesifik Esteraz (NSE), Terminal Deoksinükleotil Transferaz (TdT), Demir (Fe) boyaları kullanılmaktadır. Bunların farklı kombinasyonlarda pozitif olması FAB sınıflamasını desteklemektedir(45,46).
Tablo 3: Akut lenfoblastik lösemilerde sitokimya FAB MPO SBB AP CAE
A-EST B-EST PAS MGP Fe TdT L1 - -/np +* - +/z -/z +/- + - + L2 - -/np +* - -/z -/z +/- + - + L3 - - +* - - - +/- + - - np:nadir pozitif, *: T-ALL’de unipolar pozitif, z: zayıf,
MPO: Miyeloperoksidaz, SBB: Sudan Black Boyası, AP: Acide Phosphatase, CAE: Choloroacetate Esterase, A-EST: Alpha Napthyl Acetate Esterase, B-EST: Alpha Napthyl Butyrate Esterase, PAS:Periodic Acide Schiff, MGP: Methyl Green Pyronine, TdT: Terminal Deoxynucleotidyl Transferaz
2.2.3.İmmünfenotiplendirme
1970’li yıllarda sınıflamada immünfenotip de kullanılmaya başlanmıştır. Bazı lenfoblastların koyun eritrositleri ile rozet oluşturduğunun ve bu lösemilerin kötü seyrettiğinin saptanması ilk adımı oluşturmuş, bunu lökositlerin ortak antijeninin (cALLa = Common ALL antijen) saptanması izlemiştir. Sitoplazmik ve yüzey immünglobulinleri ve nihayet 1980’lerde antikorlardan yararlanılarak blastların kemik iliğinin hangi dizisinden farklılaştıklarını, hangi evresinden köken aldıklarını belirleyen yüzey işaretleyicileri devreye girmiştir (47). 1985’te “Lökosit Diferansiyasyon Antijenleri Çalışma Grubu” bu antijenlerin FAB alt grubu ile ilişkisini belirlemiştir. Böylece B hücre öncüleri ( pre-B, erken pre-B ) , T hücreli , B
hücreli ALL’ler ve AML’nin FAB alt grublarının tanınabilmesi mümkün olmuş, yanısıra bifenotipik, biklonal lösemi tanımları da sınıflamada yerini almıştır. “Cluster of Differantiation” (CD) adı altında standardize edilen insan lökosit tiplendirmelerinin belirlenmesi için immünoflöresans, immünositokimya ve akım sitometresi gibi yöntemler kullanılmaktadır (46).
ALL,hücre yüzeyi antijen ekspresyonuna dayanan birçok gruba ayrılabilir.En sık 5 şekil;genç pre-B-hücreli,pre B-hücreli,B-hücreli,T-hücreli ve nötr-hücreli(null-cell) ALL’dir.
ALL’li olguların yaklaşık %60’ı hücre yüzeyinde ortak ALL antijenini (CALLA) eksprese eder.CALLA(CD10),bazı normal genç lenfositlerde ve diğer hematopoetik dokular dışındaki dokularda bulunan bir glikoproteindir. CALLA(+) ALL olgularının erken pre-B-hücre farklılaşma durumunu temsil ettiği düşünülmektedir.CALLA(+) ALL’nin yaklaşık %20’sinin stoplazmik immünoglobulinleri vardır ve pre-hücreli ALL olarak adlandırılırlar. B-hücreli ALL,hücre yüzeyinde immünoglobulin bulunması ile belirlenir ve ALL olgularının %5’inden azını oluşturur.
ALL olgularının yaklaşık %20’si T-hücre fenotipindedir ve normal genç T-hücrelerinde bulunan CD5,CD3,CD2 gibi antijenleri eksprese eder.ALL olgularının yaklaşık %15’inde CALLA veya T-hücre belirteçleri yoktur ama bir B-hücre belirteci olan CD19 eksprese edilir,pro-B-ALL olarak adlandırılır.
ALL’li hastaların %25’inde lösemik hücreler myeloid belirteçleri de eksprese eder.Bu antijenlerin varlığının eskiden daha kötü prognoza sahip bir grubu tanımladığı düşünülürdü ama günümüzde agresif rejimlerle hastalık seyrinin benzer olduğu görülmektedir.(94)
2.2.4.TANI 2.2.4.1.Klinik
Lösemi tanısında da diğer hastalıklarda olduğu gibi anamnez ve fizik muayene bulguları önemli yer tutar. Klinik prezentasyon akut ya da sinsi olabilir. ALL’de klinik bulgular ve semptomlar genellikle kemik iliğinin blastlar tarafından invazyonuna bağlıdır. Bu invazyon sonucu oluşan aneminin klinik yansıması solukluk, halsizlik, çarpıntı, nefes darlığı ve ağır durumlarda kalp yetmezliği bulguları iken, lökopeniye bağlı enfeksiyon semptomları, trombositopeniye bağlı peteşi, purpura, ekimoz, mukoza ve visseral organ kanamaları gelişebilir. Kemik iliği dahil hemen tüm organlar lösemik infiltrasyona maruz kalabilirler.
ALL’de yaklaşık %30-40 oranında hepatomegali (HM) ve/veya spelomegali görülmekte, hepatosplenomegaliye (HSM) yakın oranda da lenfadenomegali (LAM) saptanmaktadır. Timusun lösemik infiltrasyonu tüm ALL vakalarında yaklaşık %10 oranında, T hücreli ALL’de %95 oranında tespit edilmiştir. Merkezi sinir sistemi (MSS) tutulumu ALL’de %5 oranında tespit edilmektedir. ALL’de testis tutulumu ise genellikle ağrısız organ büyümesi şeklinde prezente olur. Sakral sinir kökleri ya da “corpus cavernosum” ve penis dorsal venlerinin mekanik infiltrasyonuna bağlı gelişen priapism, derinin lösemik infiltrasyonu ile oluşan “lökemia cutis”, renal tutuluma bağlı gelişen hematüri, hipertansiyon, iç kulak tutulumu sonucu oluşan vertigo ve işitme
kaybı daha az veya nadir olarak görülen tablolardır (45,54,55). Süt çocukluğu lösemisinde HSM, lökositoz ve sıklıkla MSS tutulumu mevcuttur (56).
2.2.4.2.Laboratuar
Lösemi düşünülen hastada yapılması gereken basit incelemelerin başında tam kan sayımı ve periferik kan yayması gelir. Hemoglobin değeri sıklıkla hafif ya da orta derecede düşüktür. Lökosit sayısı artmış, azalmış ya da normal olabilir. Hastaların %92’sinde trombosit sayısı azalmıştır. Periferik kan yaymasında blastlar görülebilir. Kemik iliği aspirasyonu (KİA) lösemi tanısı için gereklidir ve genellikle %80-100 oranında blast infiltrasyonu tanı anında mevcuttur. Kemik iliğinde sitokimyasal boya, immünfenotiplendirme ve sitogenetik inceleme de yapılarak tanı desteklenir. Direk toraks grafisi, özellikle T hücreli lösemide sık olan mediastinal genişlemenin görüntülenmesinde, diagnostik lomber ponksiyon ise sinir sistemi tutulumunun olup olmadığının belirlenmesinde kullanılır. Ayrıca kemik grafileri, koagülasyon profili, kardiak fonksiyonların belirlenmesi, kan biyokimyası (elektrolitler, üre, kreatinin, ürik asit, fosfor, karaciğer enzimleri vs. ), enfeksiyon profili (hepatit işaretleyicileri gibi) ve immünolojik tarama (immünglobulinler gibi) tanı anında yapılması gereken diğer incelemelerdir.
2.2.5.TEDAVİ
Akut lenfoblastik lösemilerde tedavi supportif bakım, kemoterapi, kemik iliği nakli ve diğer (immünoterapi, biyoterapi) tedavi yaklaşımlarından oluşmaktadır.
A. Supportif Bakım: Lösemilerde supportif bakım genel olarak santral venöz kateter konulması, sitopenilerin replasmanı (trombosit ve eritrosit süspansiyonları), DIC profilaksisi ve tedavisi, tümör lizis sendromuna uygun medikal yaklaşım, hiperlökositozis halinde lökoferez ve/veya uygun tıbbi müdahale, enfeksiyona karşı profilaksi ve tedavi, hastaya ve ailesine psikososyal destek, erken ve geç yan etkilerin engellenmesi veya azaltılmasına yönelik yaklaşımlar şeklinde özetlenebilir (45, 55,57).
B. Kemoterapi:
İndüksiyon: Bu tedavi aşamasındaki hedef lösemik blastların kemik iliğindeki oranının %5’in altına indirilmesidir. ALL’de kemoterapi ile, 1950-1960 yıllarında; önce tek ajanla (prednizolon) %60 remisyon sağlanmış (58), daha sonra vinkristin prednizolon içeren indüksiyon protokolleri ile %85 tam remisyona ulaşılmıştır. Günümüzde yaygın olarak uygulanan dört ilaçlı indüksiyon (vinkristin, prednizolon, daunorubicin, L-asparaginaz) protokolları ile tam remisyon oranı %95 civarındadır. İndüksiyon ile birlikte MSS lösemisine yönelik profilaktik veya terapötik intratekal (İT) ilaç uygulanması da kulanılmaktadır.
Konsolidasyon: Tam remisyonu takiben uygulamaya başlanır. Rezidü blastların ortadan kaldırılması amaçlanır. İndüksiyondaki ilaçlarla çapraz direnç oluşturmayan ilaçlar kullanılarak yapılır (54). Genellikle bu dönemde MSS tutulumuna yönelik profilaktik veya terapötik radyoterapi uygulanır.
Reindüksiyon: Bazı protokollerde kısa bir ara idame periyodunu takiben indüksiyon ve konsolidasyon karışımından ibaret olan reindüksiyon fazı uygulanmaktadır.
Merkezi Sinir Sistemi Lösemisi Profilaksi veya Tedavisi:
Lösemik blastların MSS’de sekestrasyonu ve daha sonra sistemik relaps oluşumuna sebep olmasından dolayı ayrı bir tedaviye ihtiyaç vardır. MSS tutulumu yok ise profilaktik, var ise terapötik amaçla tedavi verilmektedir. ALL’li hastaların %3’ünde tanı sırasında MSS tutulumu saptanabilir. MSS profilaksisi için yüksek doz metotreksat ve/veya Ara-C veya İT metotreksat ve 1800 cGy kranial radyoterapi uygulanmalıdır. MSS tutulumunda tedavi yaklaşımı ise üçlü İT tedaviye (metotreksat, Ara-C, hidrokortizon) ek olarak 2400 cGy kranial ve 1200-1500 cGy spinal radyoterapi uygulanmaktadır(55). Bu yaklaşımlarla önceleri %50 olan MSS relaps oranı %5’lere indirilmiştir. Ayrıca bu uygulamalar genel sürvi oranını da artırmıştır.
Testiküler Lösemi Tedavisi:
Lösemide testis tutulumu testiküler büyümenin yanında testis biyopsisi yapılarak lösemik infiltrasyonun gösterilmesi ile kanıtlanmalıdır. Testis biyopsisi ile %33 oranında asemptomatik tutulum tespit edilmiş olsa da rutin yapılması önerilmemektedir. Asemptomatik vakalarda sistemik kemoterapi dışında ilave bir müdahale yapılmaz. Semptomatik ve biyopsi ile ispatlanmış hastalarda ise kemoterapiye ek olarak 1200 cGy testiküler radyoterapi önerilmektedir. İyi tedavi edilmemiş erkek çocuklarda testiküler lösemi %10 oranında geç relapslara neden olmaktadır.
İdame: Konsolidasyonu takiben rezidü blastların öldürülmesi, normal kemik iliği hematopoetik progenitörlerin korunması amacı ile uygulanır. Tedavi genellikle günlük 6-merkaptopürin ve haftalık metotreksat dozlarından oluşur. Bunlara son yıllarda aylık VCR ve kısa süreli metilprednizolon uygulanması eklenmektedir. İdame süresi 2-3 yıl olarak uygulanmaktadır. Bazı protokollarda idame tedavisi erkeklerde daha uzun tutulmaktadır (59).
Relaps Tedavisi: Lösemilerde tedavideki yetersizliğin en önemli kanıtı relaps (hastalığın tekrarlaması)’dır. ALL’li hastaların %25-30’unda relaps gelişmektedir (46,60). Relapsların %80 kadarı kemik iliği %12-16’sı MSS, %8’i testis relapsı şeklindedir. İkinci remisyon sağlanmasını ise relaps süresi, daha önceki KT’nin yoğunluğu, sekonder tedavinin tipi gibi bazı faktörler belirler. Tedavi kesildikden (6 ay) sonra oluşan geç relapslar tedavi sırasında oluşan (18 ay) relapslardan daha iyi gidişlidir (46). Relapsların 1/3’ü kemoterapi kesildikden 6 ay veya daha sonra oluşur ve yeni KT’ye cevap verir. İlk remisyon süresi de relapsta önemlidir. İlk remisyonun kısa sürdüğü hastalarda uzun süreli hastalıksız yaşam süresi %10’un altındadır(61). Relapslarda (özellikle tekrarlayan relapslarda) ilaçlara karşı direnç gelişimi tedaviye cevapta önemli rol oynar (46). Yeni tanı almış vakalarda sürvi oranları yüksek iken relaps yapmış hastalarda bu oran oldukça düşüktür. Relaps vakaları için üzerinde fikir birliği yapılan protokol ya da protokollar mevcut değildir. Genellikle hastanın önceden almış olduğu kemoterapi protokolünden daha yoğun bir protokol seçilmektedir. Günümüzde ikinci remisyonun indüksiyonu
amacıyla yüksek doz ifosfamid-etoposid(55), Modifiye ALL-REZ BFM 85 (PRD, VCR,1 g/m2 MTX, 3 g/m2 Ara-C, L-Asp.), MSKCC (3 g/m2 Ara-C, MTX, L-Asp.,VCR, PRD) gibi protokollar kullanılmaktadır (55).
C. Kemik İliği Transplantasyonu (KİT): ALL’de önceleri multipl relaps yapmış veya tedaviye dirençli hastalar KİT kapsamına alınmış ve bazılarına relaps halinde iken KİT yapılmıştır. Bu vakaların ancak %10’unda sürvi elde edilebilmiştir. Daha sonra remisyon halinde nakil tercih edilmiş, genellikle ikinci remisyonda KİT yapılmış ve sürvi oranı %27-40 olarak rapor edilmiştir. ALL’de KİT ile ikinci remisyonda sağlanan kür oranı genel olarak %30-50 oranındadır. Son yıllarda Ph+ ve t(4;11) tespit edilen vakalarda ilk remisyonda KİT yapılması tartışılmaktadır.
2.2.6.PROGNOZ
Akut Lenfoblastik Lösemi’de birçok klinik ve laboratuar özellik prognostik değer taşımakta ve nüks olasılığına, dolayısı ile remisyon sürelerine yansımaktadır. Prognostik kriterlerin incelenmesi hastalığın biyolojisinin tanınmasının yanısıra, risk gruplarının belirlenerek bunlara uygun yoğunlukta ve nitelikte tedavi yaklaşımlarının seçilmesi açısından önem taşır. Ancak, değişik araştırma gruplarınca benzer prognostik faktörler üzerinde durulsa da, bunların risk sınıflamasında kullanımı farklı olabilmekte ve ayrıca uygulanan tedaviye göre de bazı faktörlerin önemi değişebilmektedir.
Bir dönem çok önem taşıyan bazı prognostik faktörler tedavinin yoğunlaşması ile geri planda kalmış ve tedavi en önemli prognostik kriter haline gelmiştir (84,85). Diğer taraftan, risk gruplamasında kullanılan bazı özellikler örtüşmekte, örneğin belli bir sitogenetik özellik belli bir yaş grubunda yoğunlaşmaktadır (86,87). Bu nedenle bağımsız risk faktörleri elde etmek zorlaşmakta ve bir faktöre göre risk grubunu başka kriterlere göre ayrıca alt gruplara ayırmak gerekmektedir. Sonuçta oldukça karmaşık hale gelen risk sınıflamalarında merkezler ve tedaviler arası karşılaştırmayı kolaylaştıracak şekilde ortak ve aynı zamanda kesin, kolay ve hızlı elde edilen kriterler en azından ana grupların oluşturulmasında yeğlenir olmuştur.Yaş, cinsiyet ve inisyal lökosit sayısı gibi yıllar içinde önemini kaybetmeyen bazı temel prognostik faktörler halen çoğu büyük çalışma grubu tarafından risk gruplamasında kullanılmaktadır. Klasik prognostik faktörlerden bazıları tedavinin yoğunlaşması ile önemini kısmen veya tamamen kaybetmiş ve sadece belli çalışma grublarında veya sınırlı olarak risk grubu sınıflamasında yerini korumuştur. Bunlar arasında malnütrisyon, immünglobulin düzeyleri, mediastinal kitle, inisyal SSS tutulumu, inisyal hemoglobin ve trombosit değerleri, French-American-British (FAB) sınıflamasına göre morfoloji ve immünfenotip sayılabilir. Ancak zaman zaman bunlardan bazıları tekrar gündeme gelmekte ve yeni koşullarda önemi tekrar değerlendirilmektedir.
2.2.7.SİTOGENETİK
Tümör hücresindeki mitotik düzensizlikler 19. yüzyılda patologlar tarafından tanımlanmış, Von Hanseman ise bu düzensizliklerin neoplazinin orijini ile ilşkili olabileceğini belirtmiştir. Nowell ve Hungelford 1960’ta kronik miyeloid lösemide Philadelphia kromozomunu bulmuşlar, 1970’te ise bantlama
teknikleri geliştirilmiştir (62). Günümüzde genetik alanındaki yeni gelişmelerin kullanılması ile ALL’de %90’ın üzerinde hastada kromozomal anomaliler tespit edilebilmektedir. Özellikle akım sitometrisi kullanımı ile hücrelerin DNA miktarı tespit edilebilmekte ve hücrenin hangi fazda olduğu belirlenebilmektedir. Bu yöntemle belirlenen ve 50’den fazla kromozoma sahip hücrelerin bulunduğu (hiperdiploidi) ALL vakalarında prognozun daha iyi olduğu bazı merkezlerce kabul edilmektedir. Hipodiploidi yani 46’dan az kromozoma sahip ALL vakalarında ise prognoz kötüdür. Yapısal kromozom anomalileri ALL’de sık olarak bulunmakta ve vakaların yaklaşık %40’ında translokasyon tespit edilmektedir. Bu translokasyonların lösemi patogenezinde rol oynadığı ve bazılarının kötü prognozla birlikte olduğu bilinmektedir. (55,63).
2.2.7.1.Erişkin ALL Hastalarında En Sık Görülen Translokasyonlar
1) t(9;22): Nowel ve Hungerfold 1960 yılında KML hastalarında G-grubu kromozom anomalilerini tanımlamışlardır. Philadelphia kromozomu olarak adlandırılan bu yeni oluşum 9 ve 22 nolu kromozomlar arasındaki resiprokal translokasyon sonucu oluşur. Bu translokasyonun kromozom 9q34’ teki 3’ ABL gen bölgesinde gen segmentini, kromozom 22q11’ deki 5’ BCR gen segmentine ekler. Böylece hibrit BCR-ABL geni oluşur (64, 65). Bu genetik değişiklik 210 kdalton ağırlığında füzyon proteini tirozin kinaz aktivitesine sahip bir proteini kodlar. Genlerin füzyonları ile tirozin kinaz normalden daha uzun sentezlenir. Bu genin oluşturduğu protein ayrıca hemotopoetik hücrelerin değişiminde rol oynar (66, 67). BCR-ABL füzyon proteinlerin yanısıra 190-230 kd uzunluktaki ek proteinler de moleküler olarak gözlemlenmiştir.Bu translokasyon erişkin ALL hastalarında %20 oranında görülür.(68).
2) t(12;21): Yeni tanı pre B hücreli ALL’li çocuk hastalarda en sık görülen ( %25-30) kromozomal anomalidir. t(12;21) (p13;q22) , ETV/RUNX1 gen füzyonu (TEL/AML1) içerir (69,70).Erişkin ALL’de ise %4 oranında görülür(94).
ETV6 geni kemik iliği hematopoezisini sağlamak gibi bir çok yapının gelişimi için gerekli olan ETS ailesinin bir üyesidir ( 71,72 ). RUNX1 geni ise bir transkripsiyon faktörünü kodlar ve normalde tüm hematopoietik hücre serilerinde mevcuttur ( 73 ). Hem ETV6 hemde RUNX1 genleri değişik tipte insan hematolojik malignensilerinde bulunan kromozomal değişikliklere karışmışlardır ( 74,75). Bu gen mutasyonuna sahip hastalar her zaman B fenotiplidir ve çoğu 1-10 yaş arasındadır. Bu hastalarda t(9-22) ve hiperdiploidi çok nadiren görülür, bazı çalışmalarda t(12;21) li hastalarda mükemmel prognoz saptanmıştır ( 71,76,70,74,77 ).
3) t(1;19): Bu translokasyon, E2A ve PBX1 genleri arasında meydana gelir ve tüm ALL ‘lerde ikinci sıklıkta görülür ( %5 ).
E2A’nın 5’ ucu ile PBX1 arasında görülür ( 78 ). İnvitro ve invivo çalışmalarda hücre transformasyonu yeteneğine sahip bir kimerik protein oluşumuna yol açar. Pre B hücreli ALL’lerin % 25’inde ve tüm ALL’lerin % 5’inde görülür. Bu lösemi tipi klinik ve biyolojik bulguları ile iyi tanınmıştır ( 79 ).
4) t (4;11): Kötü prognozlu infant ALL’de görülür. MLL-AF4 füzyon geni mevcuttur ancak iyi anlaşılamamıştır. On iki yaş altında en sık görülen translokasyondur(81,82,83). Erişkin ALL’lerin ise sadece %5’inde görülür (68,80).
Tablo-4: Sitogenetik anormallikler Sitogenetik
Translokasyon
Moleküler Gen
Fenotip
t(9-22) (q34;q11) BCR/ABL Erken pre B hücreli t(1-19 ) (q23;p13) E2A/PBX1 Pre B hücreli
t(4-11 ) (q21;q23) MLL/AF4 Erken pre B hücreli, sıklıkla miyeloid antijen birlikteliği
t(12-21) (p13-q22) TEL/AML1 B hücreli
3.MATERYAL VE METOD
Ocak 2010 - Ağustos 2010 tarihleri arasında,Dicle Üniversitesi Tıp Fakültesi Dahiliye Anabilim Dalı / Hematoloji Bilim Dalı’na başvuran,eski ve yeni tanılı 39 erişkin ALL hastası (23 kadın,16 erkek). çalışma kapsamına alındı.
Hastalarımızda yaş,cinsiyet , tanı anındaki lökosit sayısı, hemoglobin , hematokrit, trombosit değerleri, biyokimyasal parametreleri, sedimentasyon, CRP değerleri, ekstramedullar tutulum varlığı (MSS tutulumu , testis tutulumu , mediastinal kitle,lenfadenopati), hepatosplenomegali, FAB sınıflaması, immünfenotip, boya özellikleri, aile öyküsü incelendi.
Çalışma grubundaki hastalar ALL ile uyumlu hücre morfolojisi ve yüzey antijenlerine sahipti. Tüm hastalardan anemnez alınıp FM yapıldı.Eski tanılı hastaların dosyalarından tanı anındaki lökosit sayıları ile birlikte diğer labaratuar değerlerine ulaşıldı.Yeni tanı alan hastalara da rutin labaratuar tetkikleri yapıldı (TK, BK, PY, Sedim, CRP). Tanılarının teyid edilmesi amacıyla boyama özellikleri incelendi (MPO, Sudan-Black, PAS). İmmünfenotip tayini için lenfoid işaretleyici antikorlar kullanıldı; B hücre serisi için CD10,CD19,CD20,CD22,CD24 kullanıldı.T hücre serisi içinse CD2,CD3,CD4,CD5,CD7 kullanıldı. Herhangi birinin %30’dan fazla olması pozitif olarak kabul edildi.
Tüm hastalar için, DÜTF(Dicle Üniversitesi Tıp Fakültesi) Hematoloji Laboratuarı’nda, sitogenetik inceleme amacıyla rutin olarak çalışılan translokasyonlara ( t(9;22), t(4;11), t(12;21), t(1;19)) Kantitatif PCR yöntemiyle bakıldı.
Bu yöntemde,hastadan alınan kan örneğinde lökosit sayısının 10000’in altında olmasına dikkat edildi.10000’in üzerinde olanlar izotonik sıvı ile dilue edildi.Hasta kanından,Roche Manga Pure Compact cihazında hazır izolasyon kiti ile mRNA izolasyonu yapıldı. mRNA ‘dan cDNA elde edildi.Termal Cycler cihazı kullanılarak elde edilen cDNA’dan,translokasyon parametreleri uygun primerlerle ayrı ayrı çalışıldı.Çalışmada Roche diagnostikanın Light Cycler 2.0 cihazı kullanıldı.
4.BULGULAR
Bu çalışma, DÜTF Dahiliye ABD/Hematoloji Kliniği’ne başvuran, remisyonda olan veya tedavisi devam eden, daha önce tanı konmuş veya Ocak 2010 – Ağustos 2010 tarihleri arasında yeni tanı almış olan ALL tanılı 39 erişkin hasta bulgularını içermektedir.
Çalışmada incelenen hastalardan, 23 ‘ü kadın 16 ‘sı erkek idi( %59 kadın , % 41 erkek). Hastaların yaş dağılımı 16-78 arasındaydı. Yaş ortalaması 27,5 idi (aritmetik ortalama ±standart sapma şeklinde; 27,5 ±14,7).
Hastaların ilk başvuru anında yapılan rutin fizik muayeneleri sonucunda 39 hastanın 25’inde hepatosplenomegali (%71,4), 9’unda ekstrameduller (%25,7) tutulum saptandı. Ekstrameduller tutulum; 6 hastada MSS ,1 hastada MSS ve testis, 2 hastada mediastinal tutulum şeklinde idi. Mediastinal tutulumu olan bir hastada t(9;22) kromozomal anomalisi mevcuttu. Bu hasta 56 yaşında bayan hasta idi ve indüksiyon tedavisi altında iken nötropenik ateş + sepsis nedeni ile kaybedildi.
Hastaların çok az bir kısmında (%3) aile öyküsü mevcuttu. Mesleklerine
yönelik incelemede ise birinci sırada %51,3 ile evhanımları vardı ( hastalarımızın %59’u bayandı). İkinci sırada %28,2 ile öğrenciler vardı.
Üçüncü sırada %10,3 ile esnaf vardı. Geri kalanı ise %5,1 ile aynı orana sahip işçi ve işsiz grupları idi.
Yapılan labaratuar tetkiklerinde , hastaların tanı anındaki tam kan , biyokimya , sedim ,CRP , akım sitometri (CD’ler), boya özellikleri ( PAS, MPO, SB) incelendi. Hastaların %25,7 ‘sinde tanı anındaki lökosit sayısı düşük bulunurken, % 20’si normaldi, % 54,3’ü ise yüksek lökosit sayısına sahipti. Hastaların %96,8 ‘inde anemi mevcuttu. Trombositopeni ise hastaların %85,7’sinde mevcuttu. Hastaların % 19,4 ‘ünde CRP normal iken , % 80,6 ‘sında CRP yüksek bulundu. Sedimentasyon değeri de % 23,5 oranında normal iken % 76,5 oranında yüksek bulundu. Biyokimyasal parametrelerden dikkat çekici olan sonuç LDH ‘a aitti , % 93,3 oranında yüksek LDH sonuçları mevcuttu.
Boyama özellikleri ise şöyle idi : MPO %2,6 ( +), SB %40 (+), PAS %52 (+). Kemik iliği aspirasyonunda bakılan immünofenotipleme sonuçlarına göre değerlendirilen hastalarımızın immünfenotipik olarak % 71,4 ‘ü T-hücreli , % 22,9 ‘u B-hücreli idi. 2 hasta ise bifenotipik olarak değerlendirildi ( % 5,7 ). ALL subtiplendirmesine bakıldığında da hastaların % 85,7 ‘si L2 morfolojisinde idi. % 14,3 ‘ü L1 morfolojisine sahipti. L3 subtipine ise rastlanmadı.
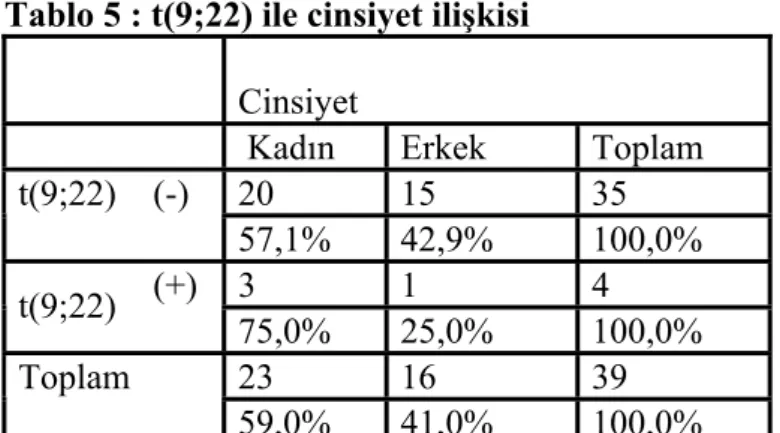
Hastaların sitogenetik incelemesi sonucunda ise 39 hastanın 4’ünde kromozomal anomali saptandı (%10,2). Bu kromozomal anomalilerin hepsi t(9;22) translokasyonu ( Philadelphia kromozomu) idi. Labaratuarımızda rutin ALL paneli kapsamında çalışılan diğer 3 translokasyon ise ( t(4-11), t(1;19), t(12;21) negatif bulundu.
Tablo 5 : t(9;22) ile cinsiyet ilişkisi
Cinsiyet
Kadın Erkek Toplam
20 15 35 t(9;22) (-) 57,1% 42,9% 100,0% 3 1 4 t(9;22) (+) 75,0% 25,0% 100,0% 23 16 39 Toplam 59,0% 41,0% 100,0%
Tablo 6 : t(9;22) ile meslek ilişkisi
Meslek Toplam
Ev
hanımı Öğrenci Esnaf İşçi İşsiz
17 10 4 2 2 35 t(9;22 (-) 48,6% 28,6% 11,4% 5,7% 5,7% 100,0% 3 1 0 0 0 4 t(9-22 (+) 75,0% 25,0% ,0% ,0% ,0% 100,0% 20 11 4 2 2 39 Toplam 51,3% 28,2% 10,3% 5,1% 5,1% 100,0%
Tablo 7 : t(9-22) ile tanı anındaki lökosit sayısı
Lökosit Toplam Düşük Normal Yüksek 9 5 17 31 t(9;22) (-) 29,0% 16,1% 54,8% 100,0% 0 2 2 4 t(9;22) (+) ,0% 50,0% 50,0% 100,0% 9 7 19 35 Toplam 25,7% 20,0% 54,3% 100,0%
Tablo 8 : t(9-22) ile anemi ilişkisi
Anemi Toplam Var Yok 30 1 31 t(9;22) (-) 96,8% 3,2% 100,0% 3 1 4 t(9;22) (+) 75,0% 25,0% 100,0% 33 2 35 Toplam 94,3% 5,7% 100,0% 26
Tablo 9 : t(9-22) ile trombositopeni ilişkisi Trombositopeni Toplam Var Yok 27 4 31 T9–22 (-) 87,1% 12,9% 100,0% 3 1 4 T9–22 (+) 75,0% 25,0% 100,0% 30 5 35 Toplam 85,7% 14,3% 100,0%
Tablo 10 : t(9;22) ile CRP ilişkisi CRP
Normal Yüksek Toplam
5 22 27 T9–22 (-) 18,5% 81,5% 100,0% 1 3 4 T9-22 (+) 25,0% 75,0% 100,0% 6 25 31 Toplam 19,4% 80,6% 100,0%
Tablo 11 : t(9-22) ile Sedimentasyon ilişkisi Sedim Toplam Normal Yüksek 7 23 30 T9-22 (-) 23,3% 76,7% 100,0% 1 3 4 T9-22 (+) 25,0% 75,0% 100,0% 8 26 34 Toplam 23,5% 76,5% 100,0% 27
Tablo 12 : t(9;22) ile ekstrameduller tutulum arasındaki ilişki Extrameduller tutulum Toplam Var Yok 7 24 31 T9-22 (-) 22,6% 77,4% 100,0% 2 2 4 T9-22 (+) 50,0% 50,0% 100,0% 9 26 35 Toplam 25,7% 74,3% 100,0% Tablo 13 : t(9;22) ile HSM* arasındaki ilişki
*HSM : Hepatosplenomegali
Tablo 14: t(9;22) ile aile öyküsü ilişkisi aile öyküsü Toplam Var Yok 1 28 29 T9-22 (-) 3,4% 96,6% 100,0% 0 4 4 T9-22 (+) ,0% 100,0% 100,0% 1 32 33 Toplam 3,0% 97,0% 100,0%
Tablo 15 : t(9;22) ile immünfenotip ilişkisi
İmmünfenotip Toplam
B-hücreli T-hücreli Bifenotipik
6 23 2 31 T9-22 (-) 19,4% 74,2% 6,5% 100,0% 2 2 0 4 T9-22 (+) 50,0% 50,0% ,0% 100,0% 8 25 2 35 Toplam 22,9% 71,4% 5,7% 100,0% HSM Toplam Var Yok 23 8 31 T9-22 (-) 74,2% 25,8% 100,0% 2 2 4 T9-22 (+) 50,0% 50,0% 100,0% 25 10 35 Toplam 71,4% 28,6% 100,0% 28
Tablo 16 : t(9;22) ile LDH* ilişkisi LDH Toplam Normal Yüksek 2 24 26 t(9;22) (-) 7,7% 92,3% 100,0% 0 4 4 t(9;22) (+) ,0% 100,0% 100,0% 2 28 30 Toplam 6,7% 93,3% 100,0% *LDH: Laktat Dehidrogenaz
Tablo17: t(9;22) ile morfolojik tip ilişkisi Subtip Toplam L1 L2 2 10 12 t(9;22) (-) 16,7% 83,3% 100,0% 0 2 2 t(9;22) (+) ,0% 100,0% 100,0% 2 12 14 Toplam 14,3% 85,7% 100,0% İstatistiki Değerlendirme:
Sürekli değişkenlerin tanımlayıcı istatistikleri için ortalama ve standart sapma , kesikli değişkenlerin tanımlayıcı istatistikleri için ise gözlem sayısı ve oranlar verildi. Kesikli değişkenler arasındaki ilişkiler çapraz tablolar haline getirilerek Khi-Kare analizleri ile değerlendirildi.
İstatistik değerlendirmede SPSS 15,0 for Windows (SPSS Inc., Chicago, IL, USA) kullanıldı. Hipotezler çift yönlü olup p≤0,05 olduğu durumda farklılık önemli olarak kabul edildi.
5.TARTIŞMA VE SONUÇ
Lösemilerin tanımlanmasından bu yana yapılan çalışmalar ve geliştirilen teknikler neticesinde hastalıkların patogenezi ciddi oranda aydınlatılmıştır. Genetik mutasyonların, kan hücrelerinin farklılaşma sürecinde hücre biyolojisini bozarak lösemiye yol açtığı düşünülmektedir. Bu mutasyonların ortaya konulması hastalığın klinik seyri ve prognozu hakkında fikir verebilmektedir.
Çalışmamızda erişkin ALL hastaları ele alınmıştır. Seçilen vakalar hastanemizde daha önce tanı konulmuş veya tezin hazırlanma sürecinde yeni tanı almış , tedavisi tamamlanmış veya devam eden , takipte olan hastalardan oluşmakta idi.Toplam 39 hasta çalışmaya alındı.Hastaların yaş aralığı 16-78 idi. Hastaların 4’ünde kromozomal anomali saptandı. Bunların tümü t(9;22) translokasyonu idi, yani Philadelphia kromozomu. Literatürde erişkin ALL vakalarının % 20-30 ‘unda pozitif olduğuna dair sonuçlar vardır (42,88,94). Bizim çalışmamızda ise %10,2 oranında Philadelphia kromozomu (+) bulunmuştur.
Philadelphia kromozomu, KML hastalarında sitogenetik açıdan tanı kriteri ve iyi prognoz belirteci iken ( 64,65,66,67), ALL hastalarında kötü prognoz belirtecidir (89). Bizim çalışmamızda da Ph (+) bulunan dört hastadan biri , 56 yaşında bayan hasta , indüksiyon tedavisi altında iken eksitus oldu. Diğer hastalardan ikisi remisyonda ve idame tedavileri devam etmektedir. Yeni tanı almış olan son hastanın ise konsolidasyon tedavisi devam etmektedir. Literatürde Ph(+) ‘liğinin tanı anındaki yüksek lökosit sayısı ile orantılı olarak arttığı yazılmıştır (90). Bizim çalışmamızda ise Ph(+) bulunan dört hastadan ikisinde tanı anında lökosit sayısı yüksek iken ikisinde normal bulundu. Diğer hastalarla karşılaştırıldığında aralarında anlamlı fark bulunmadı ( p=0,156) ( tablo 7).
ALL ‘de yaşın önemli bir prognostik faktör olduğu bilinmektedir. İleri yaş kötü prognoz sebebidir ( 84,85,86,87). Bizim çalışmamızda da erken dönemde eks olan hasta 56 yaşında bayandı ve Ph(+) idi. Ph(+) ile cinsiyet arasındaki ilişki incelendiğinde ise Ph(-) olan hastalarla arasında anlamlı bir fark bulunmadı (p=0,880). Ph (+) saptanan dört hastamızdan üçü kadın biri erkek idi. Çalışmamızda Ph(+) hastalar ile geri kalan hastalar anemi ve trombositopeni açısından karşılaştırıldı, Ph(+) olan üç hastada anemi ve trombositopeni vardı birinde yoktu. İstatistiksel olarak anlamlı fark görülmedi (p=0,534 ve p=1,000)(Tablo 8 ve 9).
Çalışmaya alınan hastalardan %80,6 ‘sında CRP yüksek bulundu, sedimantasyon değeri de % 76,5’inde yüksek bulundu. Ph (+) bulunan hastaların ise %75 ‘inde hem CRP hem de sedimentasyon yüksek bulundu(Tablo10 ve 11). İstatistiki olarak anlamlı farklılık gözlenmedi ( p=1,000 ve p=1,000). Biyokimyasal parametrelerden LDH yüksekliği dikkat çekici idi ancak Ph(+) hastalarla diğerleri arasında istatistiki analizde anlamlı fark bulunamadı (p=1,000)(Tablo16). Aile öyküsü tek bir hastamızda mevcuttu, bu hastada herhangi bir translokasyon veya kromozomal anomali saptanmadı(Tablo14).
ALL translokasyonu çalıştığımız 39 hastadan %71,4 ‘ünde HSM varken, Ph(+) olanlarda %50 HSM bulundu(Tablo13). İstatistiksel olarak anlamlı fark