ContentslistsavailableatScienceDirect
Journal
of
Molecular
Graphics
and
Modelling
jo u r n al ho me p ag e :w w w . e l s e v i e r . c o m / l o c a t e / J M G M
Discovery
of
high
affinity
ligands
for

2
-adrenergic
receptor
through
pharmacophore-based
high-throughput
virtual
screening
and
docking
Ruya
Yakar
a,
Ebru
Demet
Akten
b,∗aGraduateSchoolofComputationalBiologyandBioinformatics,KadirHasUniversity,Cibali,34083Istanbul,Turkey
bDepartmentofBioinformaticsandGenetics,FacultyofEngineeringandNaturalSciences,KadirHasUniversity,Cibali,34083Istanbul,Turkey
a
r
t
i
c
l
e
i
n
f
o
Articlehistory:
Accepted10July2014
Availableonline21July2014
Keywords: Virtualscreening Pharmacophoremodeling 2-Adrenergicreceptor Docking Scoring
a
b
s
t
r
a
c
t
Novelhighaffinitycompoundsforhuman2-adrenergicreceptor(2-AR)weresearchedamongthe cleandrug-likesubsetofZINCdatabaseconsistingof9,928,465moleculesthatsatisfytheLipinski’srule offive.Thescreeningprotocolconsistedofahigh-throughputpharmacophorescreeningfollowedbyan extensiveamountofdockingandrescoring.Thepharmacophoremodelwascomposedofkeyfeatures sharedbyallfiveinactivestatesof2-ARincomplexwithinverseagonistsandantagonists.Totestthe discriminatorypowerofthepharmacophoremodel,asmall-scalescreeningwasinitiallyperformedon adatabaseconsistingof117compoundsofwhich53antagonistsweretakenasactiveinhibitorsand 64agonistsasinactiveinhibitors.Accordingly,7.3%oftheZINCdatabasesubset(729,413compounds) satisfiedthepharmacophorerequirements,alongwith44antagonistsand17agonists.Afterwards,all thesehitcompoundsweredockedtotheinactiveapoformofthereceptorusingvariousdockingand scoringprotocols.Followingeachdockingexperiment,thebestposewasfurtherevaluatedbasedonthe existenceofkeyresiduesforantagonistbindinginitsvicinity.Afterfinalevaluationsbasedonthehuman intestinalabsorption(HIA)andthebloodbrainbarrier(BBB)penetrationproperties,62hitcompounds havebeenclusteredbasedontheirstructuralsimilarityandasaresultfourscaffoldswererevealed.Two ofthesescaffoldswerealsoobservedinthreehighaffinitycompoundswithexperimentallyknownKi values.Moreover,novelchemicalcompoundswithdistinctstructureshavebeendeterminedaspotential 2-ARdrugcandidates.
©2014ElsevierInc.Allrightsreserved.
1. Introduction
Human2-ARsbelongtothelargestsubfamilyof
G-protein-coupledreceptors(GPCRs)in thehumangenome, whichis the rhodopsinfamily.Alsoknownasseventransmembranedomain receptors(7TMreceptors), theyareembeddedinthecell mem-brane and have a crucial role in signal transduction from extracellularsidetointracellularsideinmanydifferent physiolog-icalpathways[1].GPCRsdealwithourphysiologicalresponsesto hormones,neurotransmittersandenvironmentalstimulantsand theyinitiate manysignalingpathways[2].Thus, many diseases suchashypertension,depression,asthma,cardiacdysfunction,and inflammation,arerelatedtothefunctioningofGPCRs[3],whichis amongthefourgenefamiliestargetedbymorethan50%ofdrugs onmarket[4–6].
∗ Correspondingauthorat:DepartmentofBioinformaticsandGenetics,Faculty
ofEngineeringandNaturalSciences,KadirHasUniversity,Cibali,34083Istanbul,
Turkey.Tel.:+902125336532;fax:+902125334327;mobile:+905393016544.
E-mailaddress:[email protected](E.D.Akten).
In2007,whenRasmussenandcoworkersdiscoveredthefirst X-raycrystalstructureofthehuman2-AR(PDBid:2RH1)[7],anew
gatewasopenedforcomputer-aideddrugdiscovery.Novel2-AR
inhibitorshavebeenintroducedusingstructure-basedand ligand-basedcomputationalalgorithms[8–11].Kolbetal.[9]screeneda libraryofapproximately1millioncompoundsviadockingusing theX-raystructure (PDB id:2RH1)and introducedtwenty-five novelantagonists,whichweretestedinaradioligandbindingassay. SixconfirmedhitswereidentifiedwithKivaluesrangingbetween
9nM and 3.2M. Docking-based virtualscreening experiments conductedbyTopioletal.[10,11]producednewchemicalclassesof hitsbesidesrediscoveringthewell-knownhydroxylamine chemo-type.DeGraafandRognan[12]modifiedtherotamericstatesof (Ser212)S5.43and(Ser215)S5.46withinthebindingsiteofthe firstX-raystructure,whichrepresentstheinactivestateof2-AR
andcreatedan“earlyactivated”model,whichwasfoundtobemore successfulindistinguishingpartial/fullagonistsfromdecoyligands indockingruns.Thisstudydemonstratedtheexistenceofsmallbut criticaldifferencesbetweenagonist-andantagonist-bound struc-tures.ThreeX-raycrystalstructuresof2-ARincomplexwiththree
antagonistsrevealedbyWackeretal.[13]alsodemonstratedminor http://dx.doi.org/10.1016/j.jmgm.2014.07.007
localstructuraldifferencesthatexistinthebindingpocketofthese complexes.Thedocking-basedvirtualscreeningstudyperformed byVilaretal.[14]usingtheX-raystructureof2-AR(PDBid:2RH1)
revealedthatantagonists(blockers)werepreferredoveragonists. Thiswasapromisingresultsincethestructureofthereceptorused asatargetwastheapoformofthestructureincomplexwitha par-tialinverseagonistcarazololandthusrepresentsaninactivestate. Moreover,usinganensembleofalternativeconformationsofthe receptorgeneratedtoaccountforproteinflexibility,theywereable toincreasethenumberofhitswithinthetop0.5%ofthescreened database.
Besides structure-based approaches, a ligand-based drug screening study by Tasler et al. [15] revealed a selective and potenthuman2-ARantagonist.Thescreening wasbasedona
pharmacophore alignment on known 3-adrenoceptor ligands,
whichgenerated a setof-adrenoceptorligands.Theirbinding affinitiesweremeasuredinvariousbindingassays.Uponfurther optimizationoftheseligands,aselectiveandpotenthuman2-AR
antagonistwithaKivalueof0.3nMwasintroduced.
Inourcurrent study,wepresenta virtualscreeningprotocol thatcombinespharmacophore-anddocking-basedapproachesto revealhigh-affinitycompoundsforhuman2-AR.Thenoveltyof
thisworkisthepharmacophoremodel,whichhasbeengenerated usingfivedifferentX-raycrystalstructuresof2-ARincomplex
withfivedifferentantagonists.Asoftoday,novirtualscreening studybasedonstructure-basedpharmacophoremodelinghasbeen reported.Thescreeneddatabasewasthe“cleandrug-like”subsetof ZINCdatabase[16].Adatasetconsistingof64knownagonistsand 53knownantagonistsobtainedfromGLIDAdatabase[17]wasused totestthediscriminatorypowerofthepharmacophoremodel.For thecompoundsthatsatisfiedthepharmacophorerequirements, aseriesofdocking experimentshavebeenconductedusingthe apoformoftheinactivecrystalstructure(PDBid:2RH1)asthe targetconformation.Compoundswithhighestbindingaffinities havebeenextractedandevaluatedbasedontheirpredictedADMET properties.Accordingly,atotalof62moleculeswithhighbinding affinityanddesirableADMETpropertieshavebeenextractedand werefurtherclassifiedbasedontheircommonfunctionalgroups.
2. Methods
2.1. Generationofthepharmacophoremodel
Five distinct inactive states of 2-AR wereused to create a
structure-based pharmacophore model using LigandScout soft-ware tool [18]. For each antagonist-bound complex structure extractedfrom ProteinDataBank (PDB ids:2RH1, 3D4S,3NY8, 3NY9,3NYA),apharmacophoremodelwasgenerated.Then,a so-called“shared”pharmacophoremodelthatsolelyconsistsofthe features existing in all fivemodels wasconstructed. Moreover, excludedvolumesrepresentingthestericallyoccupiedregionsby thereceptor,weretakenintoaccounttoincreasetheselectivityof themodel.
2.2. Assessmentofthepharmacophoremodel
Totestthediscriminatorypowerofthepharmacophoremodel, a database was created using 53 antagonists and 64 agonists obtainedfromGLIDAGPCR-LigandDatabase[17].Eachmolecule wasselectedbasedonitsuniquechemicalcompositiontoensure itsdistinctiveness (See supplementarymaterials, TablesS1 and S2).Thetwoantagonists,alprenololandtimolol,whichwereused toconstructthepharmacophoremodel,alsoexistedinthissmall database.LigandScoutsoftwaretoolwasusedtoscreen53 antag-onistsastheactiveligandsand64agonistsastheinactiveligands.
The maximum number of pharmacophore features that can be omittedduringscreeningwassetto2.Thehitcompoundsthat sat-isfiedthepharmacophoricrequirementswereevaluatedwithan in-housescoringfunction[18].
Toevaluatetheperformanceofthemodeltodiscriminateactive ligandsfrominactive ones,thereceiver operatingcharacteristic (ROC)curvewasconstructed.Eachpointonthecurvecorresponds tothepercentageofhitagonistsversusthepercentageofhit antag-onistswithascorevalueaboveacertainthreshold.Theso-called “modelexhaustion”orthe“cutoff”point wheretheslopeofthe ROCcurvestartstobecomelowerthan1(slopeofthediagonal line)wasdetermined.Thethresholdvaluethatcorrespondstothat “cutoff”pointwasthenusedforscreeningtheZINCdatabasewhere thehitcompoundswithascorevaluebelowthatthresholdvalue weresimplydiscarded.Theremaininghitcompoundswerefurther evaluatedthroughdockingexperiments.
2.3. ZINCdatabasetobescreened
Thesetofcompoundstobescreenedwasselectedastheclean drug-likesubsetofZINCdatabaseconsistingof9,928,465molecules as of June 2012.Thissubset wasespecially selected becauseit lacked anyaldehydesor thiols (alsocalled “yuck”compounds). Also,allthecompoundssatisfiedtheLipinski’sRuleofFive,with molecularweightlessthan500andhigherthan150,octanol–water partition coefficient smaller than 5, number of hydrogenbond donorslessthan5,numberofhydrogenbondacceptorslessthan 10.Inaddition,themaximumnumberofrotatablebondswasset to7andpolarsurfaceareawaslessthan150 ˚A2.
2.4. Evaluationsthroughdockingandscoring
Thetargetproteinwasselectedastheapoformofthe inac-tivecrystalstructureafterremovalofthepartialinverseagonist carazolol(PDBid:2RH1).Inthefirstdockingstage,GOLDdocking softwaretoolwithChemPLPscoringfunctionwasusedsinceit pro-videdtheshortestruntimewitharelativelyhigheraccuracyrate. Eachrunconsistedof10runsconfinedinasphericalregionof10 ˚A radiusinthebindingpocket.Thedockedconformationwiththe highestChemPLPscorevalue,theso-calledbestpose,wasselected andevaluatedbasedonitsneighboringresiduesinteractingwithin adistanceoflessthanorequalto5 ˚A.Thereexistsomewell-known keyresiduesforantagonistbindingpreviouslyreportedin exper-imentalstudies[13];Ser203,Ser204andSer207situatedonone sideofthebindingpocketandAsp113,Val114andAsn312onthe otherside.Accordingly,thefirstcriterionforsatisfyingthecorrect bindingmodewastointeractwithatleastoneresiduefromeach sideofthebindingpocket.Thecompoundsthatpassedthiscritical bindingtesthavebeenfurtherevaluatedbasedontheirscorevalue. Inthesecondstageofdocking,thecompoundswithChemPLP scorevaluesaboveacertainthresholdwereredockedtothesame apo form of thereceptor using AutoDock [19] and GOLD [20]. AutoDockperformed20runsforeachcompoundusing Lamarck-iangeneticalgorithm forconformationalsearchand AutoDock’s semi-empiricalscoringfunction.Dockingswereconfinedinagrid boxwithdimensionsof22.5 ˚A×22.5 ˚A×22.5 ˚Aandgridspacingof 0.375 ˚A.All20dockedconformationsfromAutoDockwerefurther evaluatedwithDSXscoringfunction[21] andtheconformation with the highest DSX score value was selected. In parallel to AutoDock,GOLDperformed10runsforeachcompoundina spheri-calregionof10 ˚AradiusinthebindingpocketusingbothGoldScore andChemScorescoringfunctions,andlikewise,theconformation withthehighestscorevaluewasselected.Consequently,foreach compound,atotalof3dockedconformations,eachwiththehighest scorevaluewasdeterminedfromDSX,GoldScoreandChemScore, respectively.
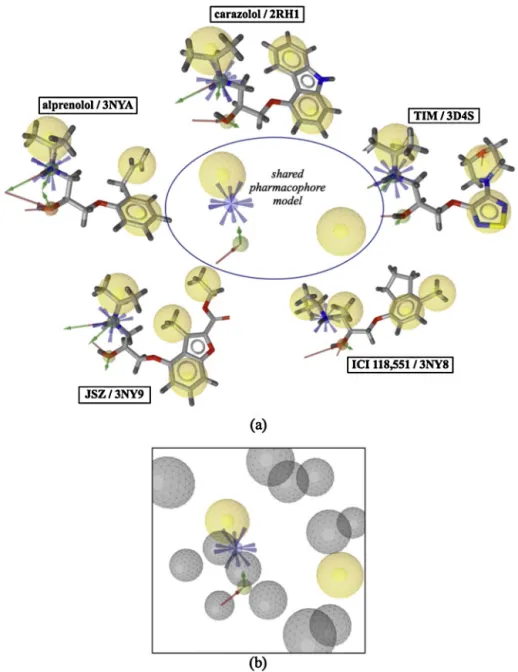
Fig.1.Pharmacophoremodelsof(a)fiveX-raycrystalstructuresofhuman2ARillustratedwithPDBidsandtheboundantagonist/inverseagonist(b)theshared
pharma-cophoremodelwithexcludedvolumesrepresentedbygrayspheres.Hydrophobicfeaturesaredepictedwithyellowspheres,hydrogenbonddonorandacceptorsbygreen
andredarrows,andpositiveionizableareabybluespheres.AllmodelsareillustratedbyLigandScoutsoftwaretool.
Eachselectedposewasfurtherevaluatedwithasecond bind-ingtestthatwasmorestringentthanthefirstone.Accordingly, theligandhastointeractwithallfourresidues,Ser203,Asp113, Asn312,andTyr316,andinaddition,witheitheroneofTyr286or Asn293[13].Thecompoundsthatfulfilledthesebindingcriteria havebeenfurtherevaluatedaccordingtotheirscorevalues.Those withascorehigherthanathresholdvaluehavebeenselectedfor thenextstagesoffiltering.Thethresholdvaluesweresetto150,77 and42forDSX,GoldScoreandChemScore,respectively.Then,all selectedcompoundsweremergedintoasinglepoolofhits;a com-poundwascountedasahitifitwasselectedinallthreedocking experiments.
Finalevaluationofthehitcompoundswasperformedusingtwo ADMETdescriptorsprovidedbyDiscoveryStudiotoolofAccelrys [22];humanintestinalabsorption(HIA)andblood brainbarrier (BBB)penetration.TheHIApropertywasdeterminedusinga pat-ternrecognitionmodelbased onpartition coefficient,logP,and polarsurfacearea,PSAandderivedfromatrainingsetof199 well-absorbedmoleculeswithactivelytransportedmoleculesremoved. TheBBBpenetration of a moleculewasdefined as theratio of
concentrationsofthecompoundonbothsidesofthemembrane afteroraladministrationandpredictedusingaregressionmodel basedon120compoundswithmeasuredpenetration.BothHIA andBBBmodelsprovide95%and99%confidenceellipses.Inthis study,thecompoundsthatfellinsidethe95%confidenceellipse foreachHIAandBBBwereproposedasplausibledrugcandidates.
3. Results
3.1. StageI.Pharmacophorescreening
The shared pharmacophore model was generated using the structuralinformationoffiveinactivecrystalstructuresin com-plexwithfivedifferentinverseagonistsand/orantagonists(PDB ids:2RH1, 3D4S,3NY8,3NY9, 3NYA).AslistedinTable S3,the pharmacophore model generated for each complex contains in averagethreeorfourhydrophobicfeatures,alsoillustratedwith yellowspheres in Fig. 1. Besides,each model holds in average threeHydrogenbonddonorsandtwoHydrogenbondacceptor fea-tures,designatedwithgreenandredarrows,respectively.Inallfive
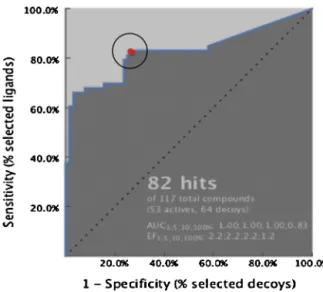
Fig.2.ReceiverOperatingCharacteristics(ROC)curveobtainedfromthescreening
of 117 molecules with known activities (53 agonists and 64 antagonists).
AUC1,5,10,100%:1.00;1.00;1.00;0.83andEF1,5,10,100%:2.2;2.2;2.2;1.2.Thereddot
illustratestheselected“modelexhaustion”orthecut-offpoint.
ligands,thereexistsonesinglepositiveionizablegrouplocatedon thebackboneNitrogenatomandrepresentedwithabluesphere. Theso-called“shared”pharmacophoremodelthatholdsthe fea-tures common to all five models consists of two hydrophobic features,oneHydrogen bonddonor,oneHydrogen bond accep-torandonepositiveionizablegroup,asdepictedatthecenterof Fig.1a.Additionally,toincreasetheselectivityofthemodel,aset of13excludedvolumespheresthatrepresentsthesterically occu-piedregionbythereceptorwasincorporated,asillustratedwith grayspheresinFig.1b.
Thesmalldatabasecomposedof53antagonistsasactiveligands and 64agonists asinactive ligandswasscreened using Ligand-Scout’s default parameters; scoring function taken as “Relative PharmacophoreFit”,thenumberofomittedfeaturessetto2,and checkexclusionvolumeturnedon.Fig.2illustratesthereceiver operationcharacteristic(ROC)curvefor82hitcompounds satisfy-ingthepharmacophorerequirementsoutof117compoundsinthe dataset.The“modelexhaustion”or“cut-off”pointonthecurve, cor-respondingto27%falsepositives(17outof64agonists)versus83% truepositives(44outof53antagonists),wasselectedtorepresent all61moleculeswithaRelativePharmacophoreFitvalueabove 0.64.Consequently,thethresholdvalueforthehigh-throughput screeningofZINCdatabasewassetto0.64.Atotalof729.413hit moleculesoutof9.928.495moleculesofthe“CleanDrug-Like” sub-setofZINCdatabasepassedthescreeningandwereselectedfor furtherevaluationthroughvariousdockingtools.
3.2. StageII.Dockingexperiments:evaluationsbasedonbinding modeandscorevalues
ThefirstdockingwasperformedusingGOLDsoftwaretool con-ductedwithChemPLP scoringfunctionsinceit provided fastest dockingrunsamong otherscoringfunctions.729,413molecules fromZINCdatabaseand61moleculesfromdatasetweredocked totheapoformoftheinactivecrystalstructure(PDBid:2RH1) withintwo months.Foreach compound,theconformationwith thehighestscorewasselectedandevaluatedbasedonthe interac-tingresiduesasdescribedinMaterialsandMethodsection(binding test#1).Atotalof610,490compoundsfromZINCdatabaseand 58molecules(41antagonistsand17agonists)havefulfilledthe binding requirementsof the first test. A threshold value of 85 wasselectedforChemPLPscoreforfurtherelimination.Thisvalue
correspondstoanenrichmentfactorof11.9fora3.2%database coveragedeterminedfrom,ER=(TP/A)/(n/N).Here,TPisthe num-beroftruepositives(knownantagonistsinourdataset),whichis 17,whereasAisthetotalnumberofantagonistsinthescreened database,whichisequalto44.Inthedenominator,nisthenumber ofselectedhitcompoundswhichis23,588andNisthetotal num-berofcompoundsinthescreeneddatabaseandisequalto729,474. Thenumberofselectedhitcompounds,23,588,issimplythesum of23,568ZINCcompounds,17antagonistsand3agonists.
Furthermore,all23,588moleculeswereredockedtothesame apoformofthereceptor(PDBid:2RH1)usingAutoDockandGOLD softwaretools.20dockedposesofAutoDockwererescoredwith DSXscoringfunctionandtheconformationwiththehighestDSX scorewasselected.Similarly,twoscoringfunctions,GoldScoreand ChemScore,wereused todeterminetheconformationwiththe highestscoreamongtendockedposesofGOLD.Eachselectedpose wasfurtherevaluatedbasedontheinteractingresidues(the sec-ondbindingtest asdescribedin Methodssection).Thenumber ofcompoundsfromZINCdatabaseandthedatasetsatisfyingthe requirementsofthesecondbindingtestisprovidedintheflowchart illustratedinFig.3.Accordingly,about10,000ZINCcompounds’ bestposefromeachthreedockingexperimentssatisfiedthesecond bindingtest.Inaddition,outof17antagonists,11to13antagonists wereamongthehitcompounds.
It isalsonoteworthythatout of17agoniststhat passedthe pharmacophorescreening,onlythreeagonistspassedtheChemPLP filterandfulfilledthesecondbindingrequirements.Sincethey rep-resentthefalsepositivesofthescreeningprotocol,theirbinding modeaswellastheirinteractingresidues,weredemonstratedin Fig.S1inordertorevealsomekeyfeaturesthatcanbeusedfor fur-therelimination.Clearly,allthreeagonistsarelargemoleculeswith atleastthreecyclicgroupsandresembletotheantagonistsandthe hitcompounds.Moreover,allthreeinteractwiththekeyresidues andwereorientedinasimilardirectioninthebindingsite(see carazololasreferenceinFigS1forcomparison).Thus,theirability topasstheinitialstagesofthescreeningtestwerenotsurprising consideringtheirmolecularsizeandtheirorientationinthe bind-ingpocket.Additionaldockingexperimentswithvariousscoring functionsbecameinevitabletoeliminatethemfromthehitlist.
Forfurtherelimination,thethresholdvaluesforDSX,GoldScore andChemScorehavebeensetto150,77,and42,respectively.The compoundswithscorevaluesbelowthesethresholdshavebeen discarded.Then,alltheremainingmoleculesweremergedintothe samepool;theywerecountedasahitiftheysatisfiedallthree thresholds.Consequently,360compoundsfromZINCdatabaseand threeantagonistsfromdatasetwereselectedashitsandfurther evaluatedbasedonADMET(Absorption,Distribution,Metabolism, ExtractionandToxicity)properties,usingDiscoveryStudiotoolby Accelrys[22].Itisnoteworthythatnoagonistwasfoundamong thehitlistsincethethreethresholdvalues(150,77,and42)have beenselectedsuchthatnoagonistwouldbeleftafterthemerge. Furthermore,approximatelytwothirdsofthehitcompoundswere eliminatedaftereachdockingevaluationbyDSX,GoldScoreand ChemScore,andwhentheresultsweremerged,onlyafew hun-dredcompoundswereleftforanalysisinmoredetailandwithina reasonabletimeframe.
3.3. StageIII.Humanintestinaladsorptionandbloodbrain barrierpredictions
The human intestinal absorption (HIA)and the blood brain barrier(BBB)penetrationwereestimatedbyADMETmoduleof Dis-coveryStudiotool. Fromdataset,onlyone antagonistmolecule, Carvedilol, was detected inside two confidence ellipses pro-vided for HIA and BBBpredictions(see Fig. S2a).On the other hand,among360compoundsfromZINCdatabase,62molecules
Fig.3. FlowchartofthescreeningprocessoftheCleanDrug-LikeZINCdatabaseandthedataset.
werefoundinsidetwo95%and99%confidenceellipsesas illus-tratedinFig.S2b.For furtheranalysis,these62moleculeswere selected and were further classified based on their chemical structure.
4. Discussion
4.1. Acloserlookatthedrugcandidatesfornovelscaffolds
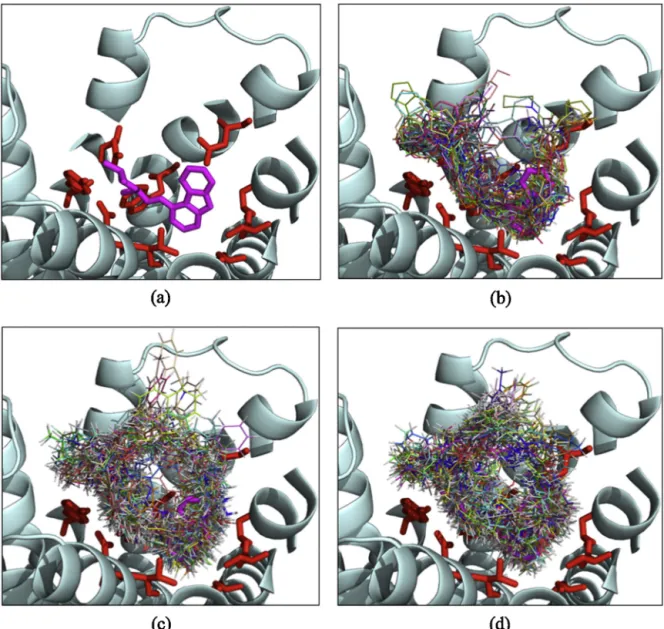
Fig.4illustrates62molecules’bestposesfromAutoDockafter beingrescoredwithDSX,ChemScoreand GoldScoretovalidate thattheybindproperlyinthebindingpocketsurroundedbykey interactingresidues.ThepartialinverseagonistCarazololinthe inactivecrystalstructure(PDBid:2RH1)wasillustratedasa ref-erencestateinallfoursnapshots.Clearly,thebestposeofeach
62compound,especiallythosegeneratedbyAutoDockruns(see Fig.4b),shareauniqueorientationthatmatcheswellwiththatof Carazolol,besidesinteractingwiththesamekeyresiduesinside thebindingpocket.Thisclearlyindicatestheuniquenessofthe bindingorientation,whicharisesfrommakingtherequiredsetof interactions.
Toidentifythechemicalgroupsontheligandthatinteractwith thetargetreceptor,the2Dchemicalstructureofall62molecules hasbeencarefullyinvestigated.Their2Drepresentationsalongside theirZINCIDandcompoundnameswereprovidedinTableS4,as asupplementarymaterial.Differentisomericformsofamolecule mightpossessverydistinctbindingmodes,whichmightleadto dif-ferentbindingaffinities.Therefore,theisomericformsofsomeof theproposedcandidatesshouldnotbediscardedandconsideredin thefuturebindingassaysaswell.7ofthosemoleculeswerefound tobetheisomerofanothercompoundinthesameset.Thus,the
Fig.4.(a)PartialinverseagonistCarazololfoundintheX-raycrystalstructure(PDBid:2RH1)demonstratedinmagentacolorandstickrepresentationforreference.Docked
posesof62moleculeswiththehighestscoreobtainedfrom(b)AutoDock-DSX,(c)GOLD/GoldScoreand(d)GOLD/ChemScoredockingruns.Keyresiduesforantagonist
bindingrepresentedinredcolorandstickrepresentation.
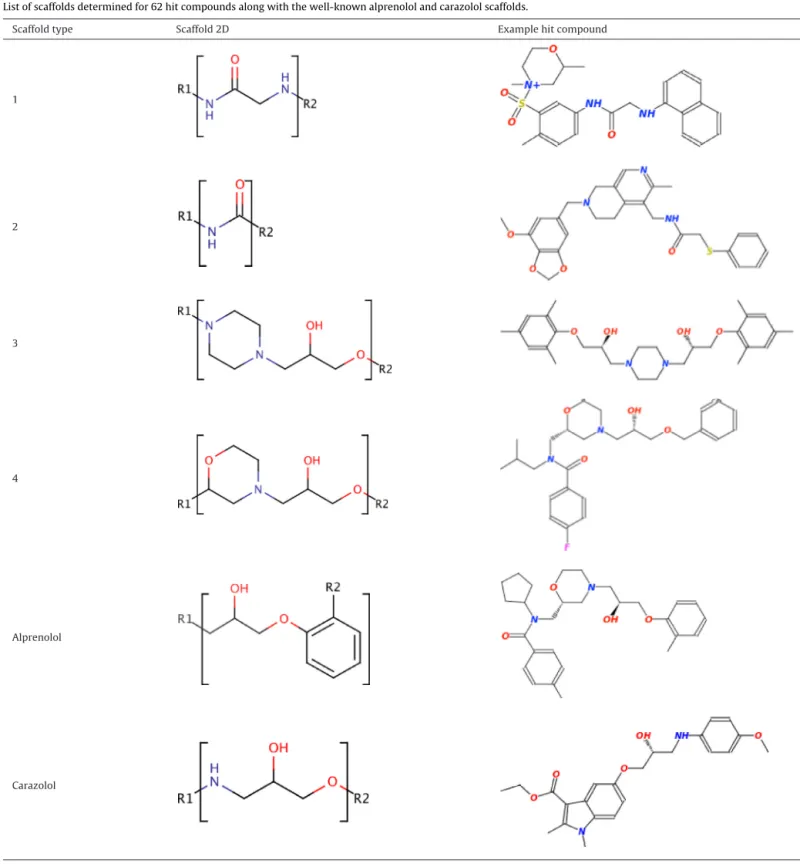
remaining55moleculeswerefurtherclassifiedaccordingtotheir fixedpartortheso-called“scaffold”.Fourdifferentscaffoldshave beendeterminedaslistedinTable1.Thenumberofcompounds thatholdsthescaffold#1,#2,#3,and#4wasfoundtobe25,10, 6,and8,respectively.Additionally,onesinglecompoundholdsthe carazololscaffoldandtheremaining5moleculeshadunique struc-turesasillustratedinFig.5.ForeachscaffoldinTable1,the2D structureofanexamplehitcompoundthatholdsthecorresponding scaffoldwasillustratedaswell.Thewell-knownclassicalscaffoldof 2ARligandscomposedofa-hydroxy-aminemotifandanether
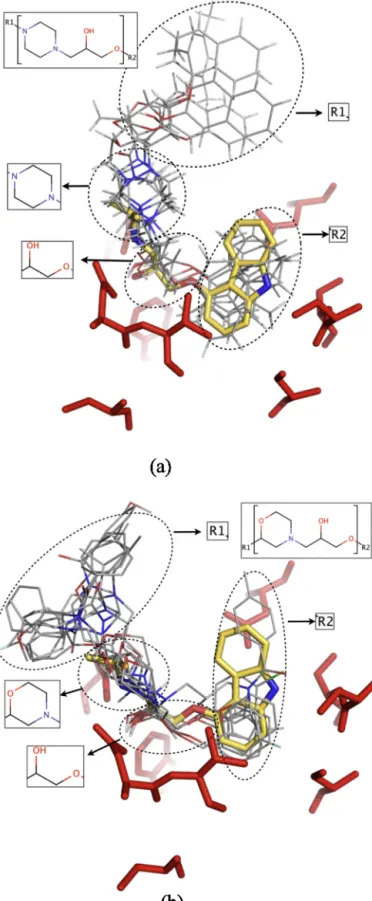
group,aslistedinTable1,holdsapartialresemblancewithscaffold #3and#4;theaminegroupintheclassicalscaffoldisreplacedbya six-memberedringinscaffold#3and#4,thatisthepiperazinewith twoaminegroupsandthemorpholinegroupwithanamineand etherfunctionalgroups,respectively.Moreover,thewidelyknown alprenololscaffoldalsoillustratedinTable1wasfoundinthreehit compoundsthatincludethescaffold#4.
Besidestheirstructuralsimilarities,thereexistsignificant over-lapsbetweenthebindingmodesofscaffold#3,#4andcarazolol. Fig.6aillustratesall6compoundsthatholdthescaffold#3withkey interactingresiduesandthecarazolol.Thesix-memberedringinall
sixcompoundscoincideswellwiththecorrespondingaminegroup ofcarazolol,makingsimilarinteractionswithAsn312illustratedin 2Dinteractionplots(supplementaryfigures,Figs.S3,S4).The oxy-genatomofcarboxamidesidegroupinAsn312makesahydrogen bondwithbackboneaminegroupincarazolol(Fig.S3),whereasit makeshydrogenbondwiththenitrogenatomintheheterocyclic ringofoneofthehitcompounds(Fig.S4).Attheoppositeside,all thehydroxyandtheethergroupsofthehitcompoundsarewell alignedwiththoseofcarazololasshowninFig.6a.Consequently, theoxygenatomofhydroxylgroupinbothcarazololandthehit compoundmakesasecondhydrogenbondwiththenitrogenatom ofcarboxamidesidegroupinAsn312(Figs.S3,S4).
Themaindifferencebetweencarazololandthehitcompounds withscaffold#3isinthehydrophobictailfacingthe transmem-branehelix5(TM5).Incarazolol,Ser203makesahydrogenbond withtheaminegroupofthecarbazoletailthatisnotpresentinthe hydrophobicheterocyclicringofanyofthehitcompounds. More-over,thepropanylendgroupofcarazololcoincideswiththelarge aromaticgroupinallsixcompounds(denotedasR1 in Fig.6a),
whichexpandstowardstheentranceofthebindingcavity, interac-tingwithLys305onTM7,Asp192onECL2,His93onTM2,Trp109on
Table1
Listofscaffoldsdeterminedfor62hitcompoundsalongwiththewell-knownalprenololandcarazololscaffolds.
Scaffoldtype Scaffold2D Examplehitcompound
1 2 3 4 Alprenolol Carazolol
TM3.Ontheotherhand,thehydrophobiccatecholaminegroupin carazololcoincideswellwiththearomaticmoietydenotedasR2in allsixcompounds,makinghydrophobicinteractionswithTyr199, Ser207,Val114andPhe290.
Fig.6billustratesall8compoundswithscaffold#4.Theonly dif-ferencebetweenscaffold#3and#4isthesix-memberedringthat hasanetherandanaminefunctionalgroupsinscaffold#4,whereas ithastwoaminegroupsinscaffold#3.Similarly,theheterocyclic ringcoincides wellwithbackbone aminegroupincarazolol,as
wellasthehydroxyandtheethergroupsthatlineupwiththose ofcarazolol.The2Dinteractionplotforoneofthehitcompound (Fig.S5)showsthehydroxygroupmakingthreehydrogenbonds withAsp113,Asn312andTyr316,whereasthehydroxygroupin carazololinteractsonlywithAsn312.
Similartohitcompoundswithscaffold#3,thereexistalarge aromatic tailthat coincide withthe short propanyl end group of carazolol. This aromatic tail is represented by R1 group in
Fig. 5. 2D representation of the five compounds with unique structures. (a) ZINC19367103; 1,4-di(4-benzyloxy-2-butynyl)piperazine hydrochloride, (b)
ZINC34691828; (2,6-dimethoxyphenyl)-[4-[[(2S)-4-[(2-hydroxyphenyl) methyl]morpholin-2-yl]methyl]piperazin-1-yl]meth, (c) ZINC40721209;
5-[(1R)-2-[4-[[2-(2-fluorophenyl)ethylamino]methyl]phenoxy]-1-hydroxy-ethyl]-1,3-dimethyl-benzimidaze, (d) ZINC66482925;
[(2S)-3-[(2S,6R)-2,6-dimethylmorpholin-4-yl]-2-hydroxy-propyl]BLAHoneand(e)ZINC67674643;3-{[7-(2,6-dimethoxyphenyl)-9-methoxy-2,3-dihydro-1,4-benzoxazepin-4(5H)-yl]methyl}-6-methyl-4H-chromen-4-one.
interactingwithTyr308andIle309onTM7,His93andIle94onTM2, Cys191andAsp192onECL2,andTrp109onTM3.Moreover,the hydrophobiccatecholaminegroupincarazolollinesupwellwith thesingle5-memberedor6-memberedcyclicringdenotedasR2in
thehitcompounds,makinghydrophobicinteractionswithSer203, Ser207,Val114andPhe290(seeFig.S5).
Structuralalignmentsofthehitcompoundsforscaffold#1and #2withcarazololwereillustratedinsupplementarymaterialFig. S6.The glycineamide group in scaffold #1 coincides well with thebackboneamine,hydroxy andether groups of carazolol.In addition,mostofthehitcompoundswerefoundtobecorrectly orientedalongthewell-knownnativestateofcarazolol.The scaf-fold#2hasthesmallestfunctionalgroupamongothers,whichis simplyamethanamidegroup.Atotalof10hitcompoundswith scaffold #2aligned tocarazololshow a satisfactoryorientation in the binding pocket, making necessary interactions with key residues.
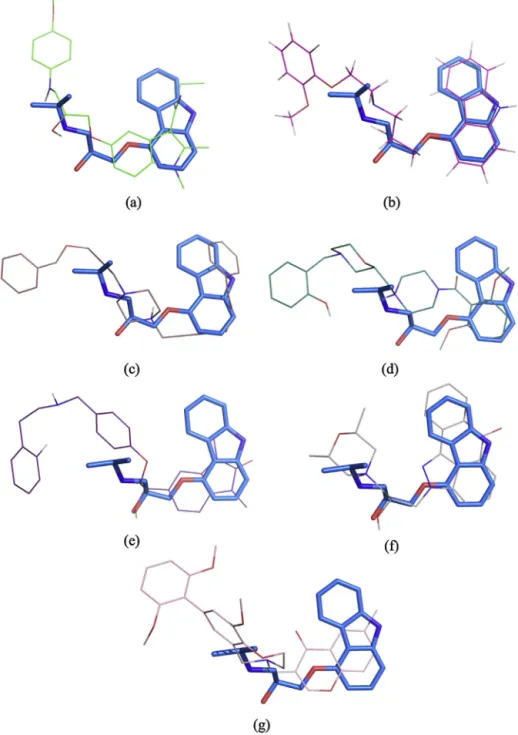
Thebestposeofthefivehitcompoundsthatdonotincorporate anyofthescaffoldslistedinTable1areillustratedwithcarazolol inFig.7.Thefirstcompound,whichistheonlyhitwithacarazolol scaffold,hasasatisfactoryorientationwithitsbackboneamine, hydroxyandethergroupsallcoincidingwellwiththoseof cara-zolol.Thesecondcompound istheantagonistcarvedilol, which isa nonselective beta-blocker (beta1,beta2),and alpha-blocker
(alpha1)andwastheonlycompoundfromdatasetthatpassedall thefilteringtests.Itisfoundtobeorientedsuitablyinthebinding pocketwithaconformationnearlymatchingthatofcarazolol.The aromaticmethoxyphenoxyringincarvedilolislineduptowards theentranceofthebindingpocket,similartohitcompoundsthat holdscaffold#3and#4.
4.2. Bindingmodesofthefivehitcompoundswithunique structures
The best pose of the five hit compounds that do not hold any of the scaffolds mentioned sofar are observed tobe cor-rectlyorientedalongsidecarazolol,asillustratedinFig.7c–g.Four of these compoundsare longer than carazololwiththeir extra aromatic tails extendingtothe entrance of thebinding pocket between TM2 and TM7, where it interacts with Gly90, His93, and Ile94onTM2,Ile309and Trp313onTM7,and, Cys191and Phe193onECL2.Consequently,thebindingcavitybecomesmore tightly packed with the ligand, which leaves a small amount ofspacefor othersmallmolecules.Anothercommonfeatureof thesefivecompoundsisthattheyallcontainatleastthree aro-maticgroups.Moreover,threeofthesegroupscontainanamine groupthatalwaysalignswellwiththebackboneaminegroupof carazolol.
Fig.6. Hitcompoundsthathold(a)thescaffold#3and(b)thescaffold#4with
keyinteractingresiduesshownwithredsticksandthecarazololinyellow.(For
interpretationofthereferencestocolourinthisfigurelegend,thereaderisreferred
tothewebversionofthisarticle.)
Thecompound#1hasasymmetricstructurewithapiperazine groupinthecenterandtwo4-benzyloxy-2butynylgroupsoneach side(seeFig.5).Thenitrogenonthepiperazinegroupcoincideswell withthebackboneaminegroupofcarazolol(seeFig.7c).Although theligandlacksanyhydrogenbondwiththereceptor,itinteracts witha totalof 21residues,includingalltheessentialones(see supplementarymaterialFig.S7).Thisis asignificantamountof interactionscomparedtocarazololsurroundedby14residuesonly (Fig.S3).Itssymmetricstructureisanothernovelfeatureforbeing acandidateforabeta-blocker.
The secondcompound hasa piperazine group attachedto a morpholinegroupatthecenter,anda2,6-dimethoxyphenyland a2-hydroxyphenylgrouponeach sideasillustratedinFig.5.It makesthreehydrogenbondswithTyr316,Asn312andAsn293, sur-roundedbyatotalof19residues(seesupplementarymaterialFig. S8).Itfitsinafavorableorientationinthebindingpocket,liningup wellwithcarazolol(seeFig.7d).Thenitrogenatomonthe piper-azinegroupplaystheroleofthebackboneaminegroupincarazolol, makinghydrogenbondwithAsn312asincarazolol.Besides,the aromatictailhydroxyphenylgroupexpandstowardstheentrance ofthebindingpocketbetweenTM2andTM7asthebenzylgroup incompound#1.
The third compound has a benzodiazole-2-one and a fluo-rophenylgrouponeachside(seeFig.5).Similartocompound#2,it iswellalignedwithcarazolol(seeFig.7e),interactswith19residues andmakesthreehydrogenbondswithSer203,Asp113andAsn312 andhasanaromatictailthatexpandstowardstheentrance(Fig. S9).Theoxygenatomonthehydroxylgroupmakestwo hydro-genbondswithbothAsp113andAsn312simultaneously.Thethird hydrogenbondisbetweentheoxygenatominthesidegroupof Ser203andtheoxygenatomonbenzodiazoleoftheligand,which coincidesinpositionwiththenitrogenofthearomaticringin cara-zolol.
Thefourthcompound hasalargearomaticmoietysimilarto thatin carazolol(seeFig.5).Theoxygenatomonthearomatic groupmakesa hydrogenbondwithSer203thatsimilarly inter-actswiththenitrogenatomofthe aromaticgroupin carazolol (seeFigs.S3andS10).Moreover,twomorehydrogenbondsare observedbetweentheligandandAsn312,inastrikinglysimilar wayasincarazolol.Unliketheotherfourcompounds,ithasashort aromatictailrepresentedbythemorpholinegroupattachedtotwo methylgroups,thatmatcheswellwiththepropanylaminetailof carazolol.Overall,italignssuitablywithcarazololasillustratedin Fig.7f.
Finally, the fifth compound is composed of three aromatic groups; a dimethoxyphenyl, a benzoxazepin and a methyl-chromene-4-onegroup(seeFig.5).Itsitsnicelyinsidethebinding pocketandislinedupwithcarazolol.Thenitrogenatomonthe benzoxazepingroupcoincidesinspacewiththebackboneamine groupincarazolol(seeFig.7g).Itinteractswith16residuesamong which Phe193onECL2,Thr110 onTM3and His93onTM2are making hydrogen bonds with the hydroxyl groups located on twoaromaticgroupsoftheligandthatexpandsupward(seeFig. S11).
4.3. Similaritiestocompoundswithknownactivities
Fig.8ashowsthe2Drepresentationofanewcompound pro-posedby Tasleret al. [15]that shows a strongbindingaffinity to human 2AR with an experimentally measured Ki value of
1.2nM.Thecompoundhasanalprenololscaffold,withR1group
asisopropyl,andR2groupasthemorpholinethatwascommonly
encounteredinthehitcompounds.Consequently,itholdsthe scaf-fold#4listedinTable1.AnotherworkbySabioetal.[11]proposed two novel compounds that alsoshow strong binding affinities withexperimentallymeasuredKi values of0.311±0.09nM and
Fig.7.Bestposesof(a)thehitcompoundwithcarazololscaffold,(b)carvediloland(c)–(g)thefivehitcompoundswithuniquestructures.Carazololrepresentedbystick
modelinblueasareference.(Forinterpretationofthereferencestocolourinthisfigurelegend,thereaderisreferredtothewebversionofthisarticle.)
57.3±1.6nM.Remarkably,bothcompoundsholdthescaffold#3 withpiperazinegroupanddiphenylmethaneasR1group,as
illus-trated in Fig. 8b and c. The R2 group has an indole in both
compounds,oneattachedtoamethylandtheothertoa carboni-trile.
Timolol and landiolol are two important beta-blockers and bothcontainmorpholinegroups[25,26].Furthermore,the activ-ity of 2 DPM derivatives (2-(3,4-dihydroxyphenyl)morpholines 3 and 4) with a morpholinic structure has been reported by Maccia et al. [27] in radioligand binding assays and func-tional tests on isolated preparations and exhibited similar adrenergic receptor activity with norepinephrine and isopre-naline. Also, piperazine group is found in antiaginal drugs, Ranolazine and Trimetazidine for the treatment of chronic
angina pectoris. Beta-blockers are also classified as antiaginal medications.
Finally,arepresentativecompoundfromeachscaffoldandthe five unique compounds have been searched as query in Drug-Bankdatabasewhichcontainsnearly7000drugentriestoidentify approveddrugmoleculesthatsharesomesimilaritieswithour pro-posedhitcompounds.ThefirstfourentriesinTable2belongto therepresentativecompoundsfromeachscaffold,whichbroadly resemble knownantagonists withthehighest Tanimoto coeffi-cientrangingbetween0.395and0.735.Ontheotherhand,thefive compoundswithuniquestructureshavethecorresponding Tani-motocoefficientbetween0.369and0.483.Especially,thetwohit compounds(IDs1and8)withTcvaluesbelow0.4presentnovel
Table2
Listofhitcompounds,theirnearestantagonistsinDrugBank[23],theiridentityandtheircorrespondingTanimotosimilarity.
ID Structure NearestantagonistsinDrugBank Identity Tc
1 Mirabegron abeta3adrenergic receptoragonist 0.395 2 Silodosin an alpha1-adrenoceptor antagonist 0.433 3 Alprenolol anadrenergic beta-antagonist 0.735 4 Levobunolol anonselective beta-adrenoceptor antagonist 0.554 5 Bisoprolol acardioselective beta1-adrenergic antagonist 0.407 6 Phenoxybenzamine an alpha-adrenergic antagonist 0.452 7 Procaterol along-acting beta2-adrenergic receptoragonist 0.483 8 Alfuzosin an alpha-adrenergic blocker 0.369 9 Silodosin an alpha1-adrenoceptor antagonist 0.418
Fig.8.2Drepresentationof(a)compound#35proposedbyTasleretal.[15],(b)compound#3and(c)compound#11proposedbySabioetal.[11].
5. Conclusions
A sharedpharmacophoremodel generated from fiveknown inactive crystal structures of human2AR was used to screen
the clean-drug like subset of ZINC database consisting of 9,928,465compoundsforthediscoveryofnovel2ARantagonists.
Pharmacophore-basedscreeningyielded729,413compoundsthat were docked tothe apo form of one of thefive inactive crys-talstructures.Followingaseriesofdocking/rescoring,a totalof 360compoundswerefoundtosatisfytherequirementsforkey residuesandscorevalues,andweresenttoADMETfiltering.62 compoundshavefulfilledtherequirementsforhumanintestinal absorption (HIA)and blood brain barrier penetration and thus wereproposedaspotentialbinders.Thesecompoundswere fur-theranalyzed and classifiedbased ontheircommonfunctional groups.Four distinctscaffoldshave beendetected.Remarkably, anovelcompoundproposedbyTasleretal.[15],possessoneof ourproposedscaffoldswithamorpholinegroup.Thiscompound wasexperimentally shown tohave a strongbinding affinityto human2AR witha Ki valueof 1.2nM.Moreover, timolol and
landiolol,twoimportantbeta-blockersbothcontainmorpholine groups.[25,26]Inaddition,Sabioet al.[11]proposedtwo novel compounds fromtheir screening studies for which the experi-mentalbindingaffinitiesweremeasuredas0.311±0.09nMand 57.3±1.6nM.Likewise,bothcompoundswerefoundtoholdone ofthefourproposedscaffoldswiththepiperazinegroup.Attheend, screeningmillionsofcompoundsthroughseveralstagesoffiltering, yielded62hitcompoundswithnoticeablestructuralsimilaritiesto thosewithstrongbindingaffinitiestestedexperimentally.In addi-tion,novelscaffoldshavebeendiscoveredwithlowsimilaritiesto anyknownapproveddrugs.Althoughtheexperimentalvalidation ofthesecompoundsarelacking,computationalmethodspredicts themasstrongbinders.Furthermore,thepharmacophoremodel, whichwasbasedonthestructureofreceptor–antagonistcomplex, increasestheprobabilityofthesecompoundstofunctionas antag-oniststhanasagonists.
Conflictofinterests
Theauthorsdeclarethattheyhavenocompetinginterests.
Acknowledgments
ThisworkhasbeenpartiallysupportedbyTheScientificand Technological Research Council of Turkey (TÜB˙ITAK, Project # 109M281)andKadirHasUniversityBAP(Project#2010-BAP-04).
AppendixA. Supplementarydata
Supplementarymaterial related tothis articlecan befound, in the online version, at http://dx.doi.org/10.1016/j.jmgm. 2014.07.007.
References
[1]P.A.Marjin,S.Verhoeven,C.deGraaf,L.Roumen,B.Vroling,B.S.Nabuurs,J.de Vlieg,P.G.Klomp,J.Chem.Inf.Model.51(2011)2277–2292.
[2]D.M.Rosenbaum,S.G.Rasmussen,B.K.Kobilka,Nature459(2009)356–363.
[3]M.O.Becker,Y.Marantz,S.Shacham,B.Inbal,A.Helfetz,O.Kalid,S.Bar-Halm, D.Warshaviak,M.Fichman,S.Noiman,Proc.Natl.Acad.Sci.U.S.A.101(2004) 11304–11309.
[4]K.Lundstrom,Curr.ProteinPept.Sci.7(2006)465–470.
[5]J.P.Overington,B.Al-Lazikani,A.L.Hopkins,Nat.Rev.Drug.Discov.5(2006) 993–996.
[6]S.Schlyer,R.Horuk,DrugDiscov.Today11(2006)481–493.
[7]S.G.F.Rasmussen,H.J. Choi,M.D.Rosenbaum,T.S.Kobilka,S.F.Thian,C.P. Edwards,M.Burghammer,R.P.V.Ratnala,R.Sanishvili,F.R.Fischetti,F.X.G. Schertler,I.W.Weis,B.K.Kobilka,Nature450(2007)383–388.
[8]M.Wada,E.Kanamori,H.Nakamura,Y.Fukunishi,J.Chem.Inf.Model.51(2011) 2398–2407.
[9]P.Kolb,D.M.Rosenbaum,J.J.Irwin,J.J.Fung,B.K.Kobilka,B.K.Shoichet,Proc. Natl.Acad.Sci.U.S.A.106(2009)6843–6848.
[10]S.Topiol,M.Sabio,Bioorg.Med.Chem.Lett.18(2008)1598–1602.
[11]M.Sabio,K.Jones,S.Topiol,Bioorg.Med.Chem.Lett.18(2008)5391–5395.
[12]C.deGraaf,D.Rognan,J.Med.Chem.51(2008)4978–4985.
[13]D.Wacker,G.Fenalti,M.A.Brown,V.Katritch,R.Abagyan,V.Cherezov,R.C. Stevens,J.Am.Chem.Soc.132(2010)11443–11445.
[14]S.Vilar,G.Ferinoa,S.S.Phatakb,B.Berka,C.N.Cavasotto,S.Costanzia,J.Mol. Graph.Model.29(2011)614–623.
[15]S.Tasler,R.Baumgartner,A.Aschenbrenner,A.Ammendola,K.Wolf,T.Wieber, J.Schachtner,M.Blisse,U.Quotschalla,P.Ney,Bioorg.Med.Chem.Lett.20 (2010)3399–3404.
[16]J.J.Irwin,K.B.Shoichet,J.Chem.Inf.Model.45(2005)177–182.
[17]Y.Okuno,J.Yang,K.Taneishi,H.Yabuuchi,G.Tsujimoto,NucleicAcidsRes.34 (2006)673–677.
[18]G.Wolber,T.Langer,J.Chem.Inf.Model.45(2005)160–169.
[19]G.M.Morris,R.Huey,W.Lindstrom,M.F.Sanner,R.K.Belew,D.S.Goodsell,A.J. Olson,J.Comput.Chem.30(2009)2785–2791.
[21]H.Gohlke,M.Hendlich,G.Klebe,J.Mol.Biol.295(2000)337–356.
[22]W.J.Egan,K.M.Merz,J.J.Baldwin,J.Med.Chem.43(2000)3867–3877.
[23]D.S.Wishart,C.Knox,A.C.Guo,S.Shrivastava,M.Hassanali,P.Stothard,Z. Chang,J.Woolsey,NucleicAcidsRes.34(Databaseissue)(2006)D668–D672.
[24]R.A. Laskowski, M.B. Swindells, J. Chem. Inf. Model. 51 (2011) 2778– 2786.
[25]J.Ogata,T.Okamoto,K.Minami,Can.J.Anaesth.50(2003)753.
[26]T.J.Zimmerman,H.E.Kaufman,Arch.Ophthalmol.95(1977)601–604.
[27]B.Macchia,A.Balsamo,M.C.Breschi,A.Lapucci,A.Lucacchini,F.Macchia,C. Manera,A.Martinelli,C.Martini,E.Martinotti,S.Nencetti,J.Med.Chem.35 (1992)1009–1018.


![Fig. 8. 2D representation of (a) compound #35 proposed by Tasler et al. [15] , (b) compound #3 and (c) compound #11 proposed by Sabio et al](https://thumb-eu.123doks.com/thumbv2/9libnet/4321944.70828/12.918.142.787.86.429/representation-compound-proposed-tasler-compound-compound-proposed-sabio.webp)
