Near-IR absorbing BODIPY derivatives as glutathione-activated photosensitizers for selective photodynamic action
Tam metin
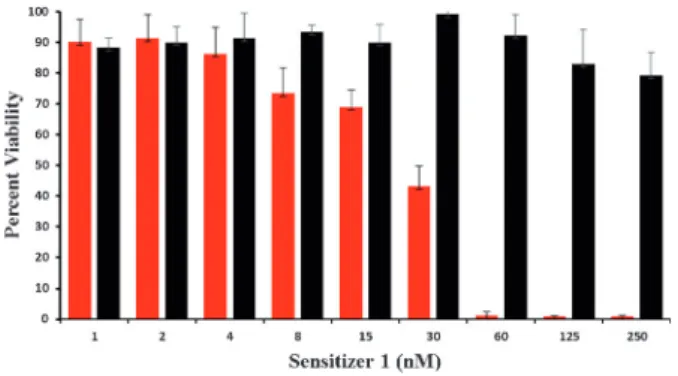
Şekil
![Table 1. IC 50 values of sensitizers with the HCT116 cell line. [a]](https://thumb-eu.123doks.com/thumbv2/9libnet/5899583.122068/3.892.460.820.132.289/table-ic-values-sensitizers-hct-cell-line.webp)
Benzer Belgeler
The power capacity of the hybrid diesel-solar PV microgrid will suffice the power demand of Tablas Island until 2021only based on forecast data considering the
· Yerel çevreye ilişkin hazırlanmış çeşitli haritalara bakarak beşeri özellikler hakkında farklı çıkarımlarda (örneğin, ana ulaşım güzergahları, kentsel
We elaborate on several solution approaches, namely object-oriented design approach, aspect-oriented design approach, and composition of events and actions approach to tackle
Chen and Choi [5] gave two algorithms and one of them bounds the load by 4L using at most 4S storage space, where L and S (defined in Section 2) are commonly used as the trivial
In this paper, we present an algebraic description of the aliasing phenomena evident in the linear sampling pro- cess of multidimensional periodic band limited signals.. Opposed to
Keywords: Greek –Turkish relations; peace and conflict studies; interactive con flict resolution; peace education; empathy;
In 1978, Kunt created the first scientific journal on sig- nal processing—entitled Signal Processing—and worked as its editor-in-chief till 2006.. In parallel with the journal,
Abone veri tabanlarının yanı sıra araştırıcıların farklı veri tabanlarını tanımaları ve elektronik kaynak koleksiyonu oluşumuna katkı sağlamaları amacıyla, her
