Roles of senescence escape and epigenetic modifications in liver cancer
Tam metin
Şekil
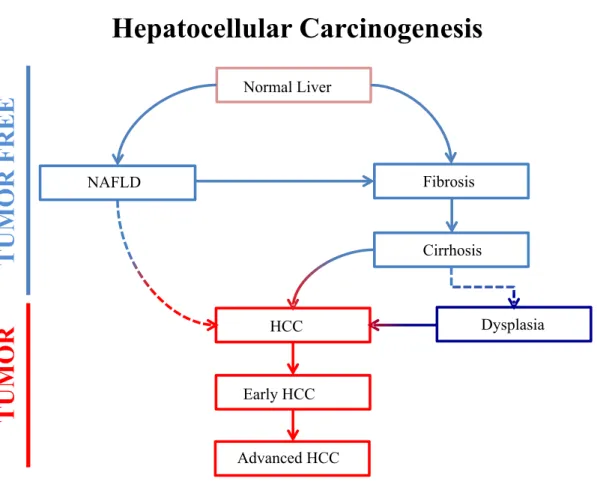
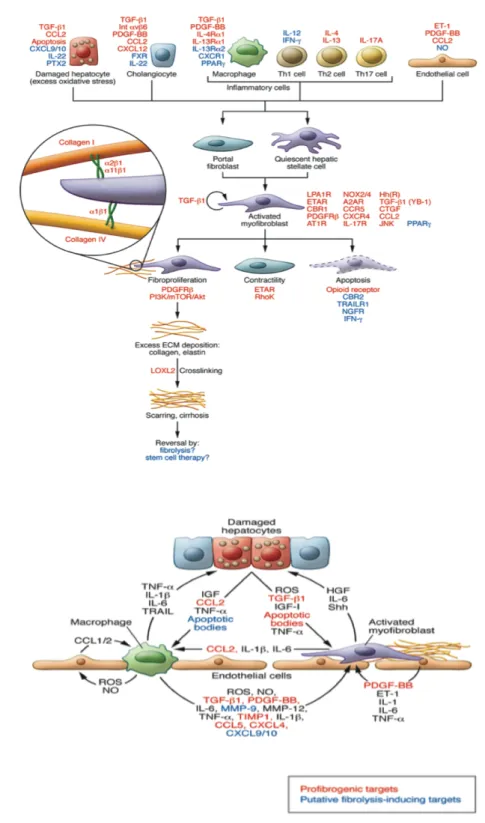

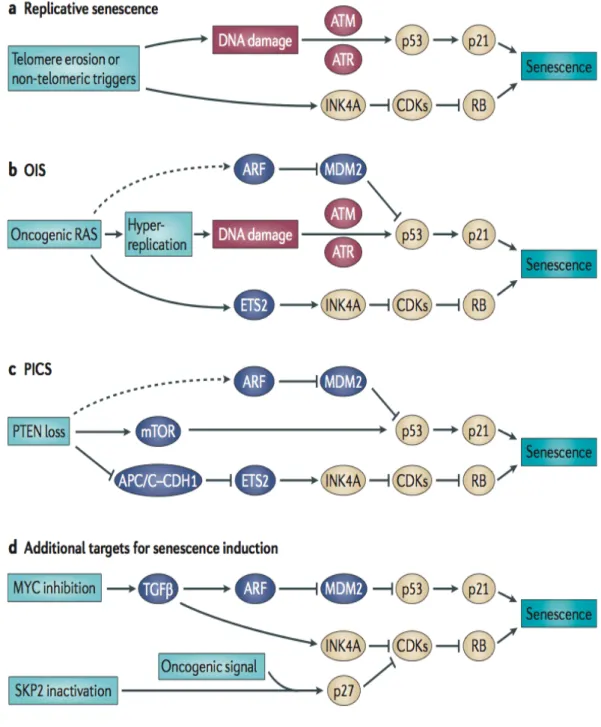
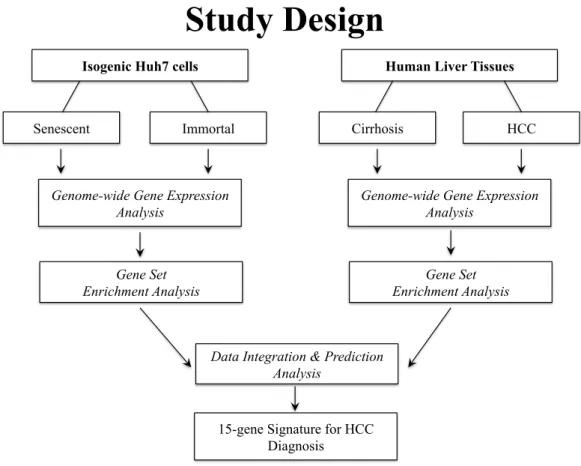


Benzer Belgeler
These charged particles are the ones that screen and shield the applied potential; therefore, if the tendency to form pairs is high (i.e., if the association constant is high),
As a result, af- ter contact of the catalysts with methane at 573–623 K in the absence of gaseous oxygen, products of partial oxidation (adsorbed formate species and formic acid)
A similar construction of non-singular projective curves with a lot of k' -rational points based on the use of fibre products of some Artin-Schreier curves was
Chap- ter 3 investigates the code design for wireless communication systems with joint RF energy and information transfer, where we propose a coding scheme based on a
As an application, we list the deformation families and compute the fundamental groups of all irreducible maximizing simple sextics with a type D singular point.. We attempt to
3 demonstrates the PDF of the LLRs corresponding to the optimized degree distributions in [2] for a two-user GMAC with equal channel gains computed via different methods.. It can
Although various noble metal (e.g. Pt, Pd and Au) structures have been synthesized by hard- and soft-- templating methods, mesoporous rhodium (Rh) nanoparticles have never
The former interaction indicates that increases of the M/T contrast ratio had divergent effects on target brightness and contour visibilities; i.e., the enhancement effect increased
