Tümör nekroz faktörü reseptör 1’in (TNFR1) sinyal iletim mekanizmasının araştırılması
Tam metin
Şekil
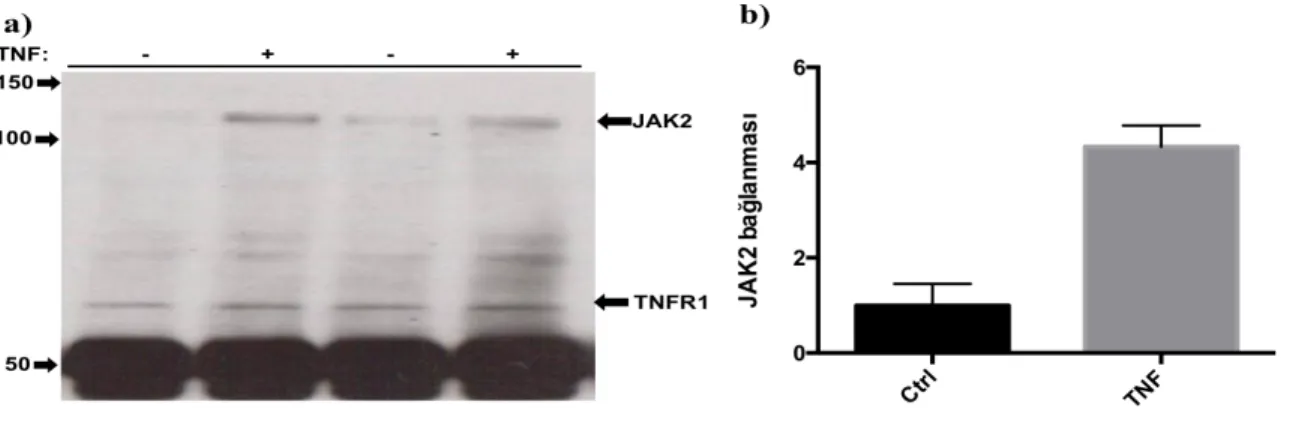
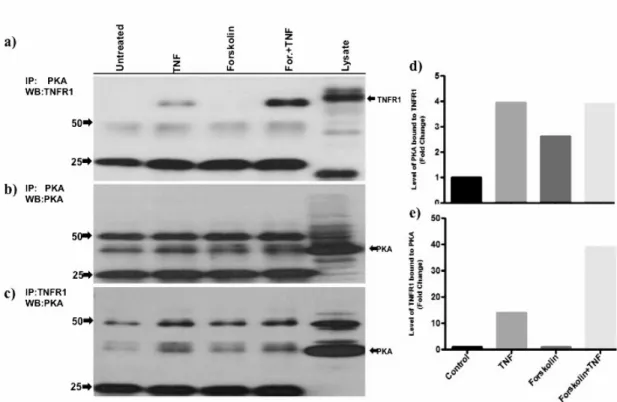








Benzer Belgeler
Ulnar arterin yokluğunda önkol dolaşımını radial ve interosseöz arterler ya da bizim olgumuzda olduğu gibi büyük bir median arter kompanse etmektedir..
Ferroşelataz enzimleridir. ALA-dehidrataz’ın inhibisyonu sonucunda -ALA → PBG’ye dönüşemez. Ferroşelatazın inhibisyonu ile sitoplazmadaki Fe +2 iyonu
Figure 2.5: The simulink diagram of the 'Hybrid Structure' block for compass gait model.. 2.4
Lenfositler, endotel ve düz kas hücreleri de TNF-a salgılayan diğer kay- naklardır (3,l5)_ Reperfüzyon zedelenmesinde önemli bir faktör olan ca+ 2 da makrofajlardan
gilloz oluşturulan sıçanların akciğer doku örneklerinde kontrol grubuna göre istatistiksel olarak anlamlı düzeyde yüksek IL-10 ve TNF- α mRNA ifadelenmesi gösterilmiş
Bu çalışmada da Aşağı Seyhan Nehri su kalitesinin, debinin maksimum olduğu yağışlı dönemlerde her üç yıl için de noktasal kirlilik kaynaklarından fazla etkilenmediği
daki bir diğer çalışma sonucuna göre prekor mutas- yonları HBeAg pozitif örneklerde %18, anti HBe pozitif örneklerde %31 iken, kor promoter mutasyon- larında bu oran
İngiliz Romatizma Birliği (BSR) yönergeleri, altta yatan ya da daha önce mevcut olan akciğer hastalığı olan hastalarda TNFİ'nin (özellikle infliximab)
