1521-0103/347/1/47–56$25.00 http://dx.doi.org/10.1124/jpet.113.206243 THEJOURNAL OFPHARMACOLOGY ANDEXPERIMENTALTHERAPEUTICS J Pharmacol Exp Ther 347:47–56, October 2013 Copyrightª 2013 by The American Society for Pharmacology and Experimental Therapeutics
Intracellular Transactivation of Epidermal Growth Factor
Receptor by
a
1A
-Adrenoceptor Is Mediated by
Phosphatidylinositol 3-Kinase Independently of Activation of
Extracellular Signal Regulated Kinases 1/2 and Serine-Threonine
Kinases in Chinese Hamster Ovary Cells
s
Nadir Ulu,
1Robert H. Henning, Sahika Guner, Teuta Zoto, Basak Duman-Dalkilic,
Marry Duin, and Hakan Gurdal
Department of Clinical Pharmacology, University Medical Centre Groningen, University of Groningen, Groningen, The
Netherlands (N.U., R.H.H., M.D.); Department of Medical Pharmacology, Faculty of Medicine, University of Ufuk, Ankara, Turkey (S.G.); and Department of Medical Pharmacology, Faculty of Medicine, University of Ankara, Ankara, Turkey (T.Z., B.D., H.G.)
Received May 3, 2013; accepted July 30, 2013
ABSTRACT
Transactivation of epidermal growth factor receptor (EGFR) bya1
-adrenoceptor (a1-AR) is implicated in contraction and hypertrophy
of vascular smooth muscle (VSM). We examine whether alla1-AR
subtypes transactivate EGFR and explore the mechanism of transactivation. Chinese hamster ovary (CHO) cells stably express-ing one subtype ofa1-AR were transiently transfected with EGFR.
The transactivation mechanism was examined both by coex-pression of a chimeric erythropoietin (EPO)-EGFR with an extra-cellular EPO and intraextra-cellular EGFR domain, and by pharmacologic inhibition of external and internal signaling routes. All threea1-AR
subtypes transactivated EGFR, which was dependent on the increase in intracellular calcium. The EGFR kinase inhibitor AG1478 [4-(39-chloroanilino)-6,7-dimethoxyquinazoline] abro-gateda1A-AR anda1D-AR induced phosphorylation of EGFR,
but both the inhibition of matrix metalloproteinases by GM6001 [(R)-N4-hydroxy-N1
-[(S)-2-(1H-indol-3-yl)-1-methylcarbamoyl-ethyl]-2-isobutyl-succinamide] or blockade of EGFR by cetuximab did not. Stimulation ofa1A-AR anda1D-AR also induced
phos-phorylation of EPO-EGFR chimeric receptors. Moreover,a1A-AR
stimulation enhanced phosphorylation of extracellular signal regulated kinase (ERK) 1/2 and serine-threonine kinases (Akt), which were both unaffected by AG1478, indicating that ERK1/2 and Akt phosphorylation is independent of EGFR transacti-vation. Accordingly, inhibitors of ERK1/2 or Akt did not influence thea1A-AR–mediated EGFR transactivation. Inhibition of calcium/
calmodulin-dependent kinase II (CaMKII), phosphatidylinositol 3-kinase (PI3K), and Src, however, did block EGFR transactivation bya1A-AR anda1D-AR. These findings demonstrate that alla1-AR
subtypes transactivate EGFR, which is dependent on an intracellular signaling route involving an increase in calcium and activation of CaMKII, PI3K, and Src, but not the of ERK1/2 and Akt pathways.
Introduction
The major importance of cardiovascular (CV) disease has generated interest in the factors that induce proliferation, hypertrophy, and contraction of vascular smooth muscle cells (VSMC) and cardiomyocytes. Prolonged stimulation of a1-adrenoceptors (a1-ARs) by catecholamines induces
hyper-trophy of VSMC (Chen et al., 1995) and cardiomyocytes (Barki-Harrington et al., 2004) and constitutes an important
This study was supported by a research grant from the Scientific and Technological Research Council of Turkey [Grant SBAG-109S125].
This work was presented as a poster as follows: Ulu N, Henning RH, Dalkilic B, Duin M, Guner S, Ugur M, Zoto T, and Gurdal H (2011)a1-Adrenoceptor subtype specificity of catecholamine induced transactivation of EGF receptor in CHO cells [poster number P007]. British Pharmacological Society Winter Meeting; 2011 Dec 13–25; London.
1
Current affiliation: Celgene Ilac Pazarlama ve Ticaret Ltd., Sti., Istanbul, Turkey.
dx.doi.org/10.1124/jpet.113.206243.
s This article has supplemental material available at jpet.aspetjournals.org.
ABBREVIATIONS: AG1478, 4-(39-chloroanilino)-6,7-dimethoxyquinazoline; Akt, serine-threonine kinases; A7R5, rat smooth muscle embryonic aorta; a1-AR, a1-adrenoceptor; Ca
21/CaM, calcium/calmodulin; CaMKII, calcium/calmodulin-dependent kinase II; CHO, Chinese hamster ovary; CV, cardiovascular; DMEM, Dulbecco’s modified Eagle’s medium; EGF, epidermal growth factor; EGFR, epidermal growth factor receptor; EPO, erythropoietin; EPOR, erythropoietin receptor; ERK1/2, extracellular signal regulated kinases; GM6001, (R)-N4-hydroxy-N1-[(S)-2-(1H-indol-3-yl)-1-methylcarbamoyl-ethyl]-2-isobutyl-succinamide; GPCR, G-protein–coupled receptor; KN93, N-[2-[N-(4-chlorocinnamyl)-N-methylaminomethyl]phenyl]-N-(2-hydroxyethyl)-4-methoxybenzenesulfonamide phosphate salt; LY294002, 2-(4-morpholinyl)-8-phenyl-1(4H)-benzopyran-4-one hydrochloride; MMP, matrix metalloproteinase; MK2206, 8-(4-(1-amino-cyclobutyl)phenyl)-9-phenyl-[1,2,4]triazolo[3,4-f][1,6]naphthyridin-3(2H)-one; pCamKII, phosphorylated CamKII; pEGFR, phosphorylated EGFR; pEPOR, phosphorylated EPOR; pErk1/2, phosphorylated ERK1/2; pPI3K, phosphorylated PI3K; PD98059, 2-(2-amino-3-methoxyphenyl)-4H-1-benzopyran-4-one; PE, phenylephrine; PI3K, phosphatidylinositol 3-kinase; PKI-166, 4-[4-[[(1R)-1-phenylethyl]amino]-7H-pyrrolo[2,3-d]pyrimidin-6-yl]phenol; PP2, 4-amino-3-(4-chlorophenyl)-1-(t-butyl)-1H-pyrazolo[3,4-d]pyrimidine; VSMC, vascular smooth muscle cells; W7, N-(6-aminohexyl)-5-chloro-1-naphthalenesulfonamide hydrochloride.
47 http://jpet.aspetjournals.org/content/suppl/2013/07/31/jpet.113.206243.DC1
at ASPET Journals on August 1, 2018
jpet.aspetjournals.org
risk factor for CV diseases. In this process, transactivation of epidermal growth factor receptor (EGFR) bya1-ARs has been
implicated as a major pathway involved in catecholamine-mediated CV hypertrophy (Asakura et al., 2002; Zhang et al., 2004; Li et al., 2011).
We recently demonstrated that transactivation of EGFR in VSMC also contributes toa1-AR–mediated vascular
contrac-tion. In that study, a1-AR stimulation resulted in EGFR
phosphorylation in rat aorta, while the EGFR kinase inhibitors AG1478 [4-(39-chloroanilino)-6,7-dimethoxyquinazoline] and DAPH (4,5-dianilinophthalimide) concentration-dependently attenuated phenylephrine (PE)-induced contractile responses (Ulu et al., 2010). Moreover, in our recent study, EGFR kinase inhibitor PKI-166 [4-[4-[[(1R)-1-phenylethyl]amino]-7H-pyrrolo [2,3-d]pyrimidin-6-yl]phenol] attenuated the progression of hypertension and maintained cardiac function in the hyper-tensive chronic kidney disease rat model (Ulu et al., 2013). The induction of vascular contraction and cellular growth and proliferation bya1-AR involves the activation of complex
signaling pathways including phospholipase C, protein kinase C, calcium/calmodulin-dependent kinase II (CaMKII), phosphatidylinositol 3-kinase (PI3K), Src, mitogen-activated protein kinases, extracellular signal regulated kinases (ERK1/2), and serine-threonine kinases (Akt) (Wu et al., 1992; Xiao et al., 2001; Illario et al., 2003; Koshimizu et al., 2003; Cipolletta et al., 2010; Haba et al., 2010). Modulation of all these intracellular signaling molecules may, at least in part, be dependent on transactivation of EGFR bya1-AR.
Previous studies have suggested that G-protein–coupled receptors (GPCRs) induce transactivation of EGFR by an extracellular route involving matrix metalloproteinase (MMP)-dependent shedding of growth-factor-like substances, for example, heparin-binding EGF-like growth factor (HB-EGF) (Prenzel et al., 1999; Zhang et al., 2004). However, in our recent study in rat aorta, we observed a1-AR–mediated
transactivation of EGFR to be independent from MMPs, which suggests that it depends on an intracellular route (Ulu et al., 2010). Therefore, in this study, we address whether an intracellular or extracellular signaling route is involved in the transactivation of EGFR by examininga1A
-AR–induced phosphorylation of a chimeric erythropoietin re-ceptor (EPOR)/EGFR in Chinese hamster ovary (CHO) cells. In addition, we used pharmacological tools interfering with extra-cellular and intraextra-cellular sites of EGFR or modulating specific intracellular signaling pathways. To conclusively determine whether all threea1-AR subtypes can transactivate EGFR, we
investigated transactivation of EGFR in CHO cells stably expressing a single subtype ofa1-AR. As ERK1/2 and Akt
acti-vation by GPCRs are often thought to originate from cross-talk with classic receptor tyrosine kinases (such as protein kinase C and EGFR) or effectors (such as arrestins) (Luttrell, 2005; Engelhardt, 2007), we also explored whethera1A-AR–dependent
activation of ERK1/2 and Akt requires EGFR transactivation.
Materials and Methods
Cell Culture. A7R5 (rat smooth muscle embryonic aorta) cells were grown in 75-cm2 nontreated cell culture flasks (Corning, Tewksbury, MA) in Dulbecco’s modified Eagle’s medium (DMEM) (GIBCO/Invitrogen, Carlsbad, CA) enriched with 10% fetal bovine serum and 1% penicillin/streptomycin. Before the experiments, the
cells were trypsinized and replated at 50% confluence on six-well plates. Stimulations were performed on day 3 after 24 hours of serum starvation.
CHO K1 cells lacking intrinsica1-AR were stably transfected with a plasmid containing one of the human a1-AR subtypes: a1A-AR, a1B-AR, ora1D-AR. They were grown in 75-cm2nontreated cell culture flasks in DMEM/Ham’s F-12 medium (DMEM/F-12) enriched with 10% fetal bovine serum, 1% penicillin/streptomycin, and 200mg/ml Geneticin (G418; Invitrogen, Carlsbad, CA). For each experiment, 104 cells were plated in each well of a six-well plate and grown for 2 days to reach a confluence of 35–40%. Subsequently, the cells were transiently transfected with the pBabe-puro plasmid (1mg) contain-ing human EGFR sequence (AddGene, Cambridge, MA). Transfection was performed with Lipofectamine 2000 (Invitrogen) at a concen-tration of 2.5ml/well according to the protocol. The next day, the cells were serum-starved for 24 hours before they were used in experiments.
Intracellular Calcium Measurements. PE-mediated calcium (Ca21) responses were measured in nontransfected and EGFR-transfected CHO cells stably EGFR-transfected with one of the a1-AR subtypes. Intracellular Ca21was measured by fura-2 fluorescence at room temperature (216 2°C). Cells were incubated for 50 minutes with 1mM fura-2-AM (Sigma-Aldrich, St. Louis, MO) in HEPES buffer (10 mM HEPES, 120 mM NaCl, 5 mM KCl, 1.5 mM KH2PO4, 1 mM MgCl2×6H2O, 5 mM NaHCO3and 1.5 mM CaCl2; pH 7.4) for loading and then washed with fresh HEPES buffer. Fluorescence was recorded using a PTI Ratiomaster microspectrophotometer and FELIX software (Photon Technology International, Inc., Birmingham, NJ).
Stimulation Experiments with A7R5 and CHO Cells. A7R5 cells were pretreated with 10mM EGFR inhibitor AG1478 or 1 mM prazosin for 30 minutes and then incubated with 10mM PE (for 5 or 10 minutes) or 1 nM epidermal growth factor (EGF; positive control, for 10 minutes) after 24 hours of serum starvation. CHO cells were pretreated for 30 minutes with one of these inhibitors: 10mM EGFR kinase inhibitor AG1478 (Sigma-Aldrich), 10 mM PI3K inhibitor LY294002 [2-(4-morpholinyl)-8-phenyl-1(4H)-benzopyran-4-one hy-drochloride] (Sigma-Aldrich), 10mM Src inhibitor PP2 [4-amino-3-(4-chlorophenyl)-1-(t-butyl)-1H-pyrazolo[3,4-d]pyrimidine] (Sigma-Aldrich), 100 mM Ca21/CaM inhibitor W7 [N-(6-aminohexyl)-5-chloro-1-naphthalenesulfonamide hydrochloride] (Tocris Bioscience, Bristol, United Kingdom), 10 mM CaMKII inhibitor KN93 (N-[2-[N-(4- chlorocinnamyl)-N-methylaminomethyl]phenyl]-N-(2-hydroxyethyl)-4-methoxybenzenesulfonamide phosphate salt) (Tocris Bioscience), 10mM broad spectrum MMP inhibitor GM6001 [(R)-N4-hydroxy-N1 -[(S)-2-(1H-indol-3-yl)-1-methylcarbamoyl-ethyl]-2-isobutyl-succinamide] (Calbiochem, Merck KGaA, Darmstadt, Germany), 10mM ERK1/2 inhibitor PD98059 [2-(2-amino-3-methoxyphenyl)-4H-1-benzopyran-4-one] (Sigma-Aldrich), and 1 mM Akt inhibitor MK2206 [8-(4-(1-amino- cyclobutyl)phenyl)-9-phenyl-[1,2,4]triazolo[3,4-f][1,6]naphthyridin-3(2H)-one] (Selleckhem, Houston, TX). Pretreatment with the anti-EGFR monoclonal antibody cetuximab (Merck Serono, Geneva, Switzerland) was performed for 2 hours at a concentration of 5mM. Stimulation with 10mM PE or 1 nM EGF was terminated after 10 minutes of incubation by removing the media, placing the plates on ice, and adding the ho-mogenization buffer. Each experiment was performed in triplicate and repeated as specified under Results.
EPOR-EGFR Chimeric Receptor. On the 3rd day after plating, CHO cells containing thea1A-AR subtype were transfected with the pcDNA3 plasmid containing the EPOR-EGFR chimeric receptor, a generous gift of Dr. Hong-Jian Zhu (Ludwig Institute for Cancer Research, Melbourne, VIC, Australia). The chimera embodied the extracellular domain of the EPOR fused to the transmembrane and intracellular domains of the EGFR (Zhu et al., 2003). On the 5th day, 24-hour serum-starved cells were stimulated with 10mM PE or 100 IU×ml21EPO (positive control) for 10 minutes. Each experiment was performed in triplicate and was repeated as specified under Results.
at ASPET Journals on August 1, 2018
jpet.aspetjournals.org
Protein Isolation and Western Blotting. Cells were immedi-ately put on ice after stimulation, and 100ml of buffer containing 1% Nonidet P40, 0.02 M sodium vanadate, protease inhibitor (Roche Molecular Diagnostics, Mannheim, Germany) in phosphate-buffered saline was added. After homogenization, cells were incubated for 15 minutes and centrifuged at 5000g for 5 minutes at 4°C. Supernatant was collected and stored at280°C.
We performed electrophoresis on newly cast 4–7% SDS-polyacrylamide gels and then transferred them onto polyvinylidene difluoride (PVDF) membranes. The membranes were incubated with the appropriate primary antibodies overnight, washed three times for 10 minutes with Tris-buffered saline and 0.2% Tween 20 and incubated for 1 hour with the secondary antibody. After subsequent washes, the membranes were soaked in Luminol (Santa Cruz Biotechnology, Santa Cruz, CA) and imaged on Kodak film.
We used the following antibodies (with the concentration for Western blot analysis): anti-EGFR (1:500), anti-Src (1:1000), phos-phorylated EGFR 1173 (pEGFR; 1:500), phosphos-phorylated EPOR (pEPOR; 1:500), phosphorylated Erk1/2 (pErk1/2; 1:5000), phosphor-ylated PI3K (pPI3K; 1:400), phosphorphosphor-ylated CamKII (pCamKII; 1: 2000),b-actin (1:5000), goat anti-rabbit (1:10,000), rabbit anti-goat (1: 10,000), and donkey anti-mouse (1:10,000) from Santa Cruz Bio-technology, and phosphorylated Akt (pAkt; 1:1000) and phosphory-lated Src 416 (pSrc 416; 1:1000) from Cell Signaling Technology (Danvers, MA). When needed, membranes were stripped with 2% SDS, 0.7% mercaptoethanol in 10 ml of phosphate-buffered saline, and then incubated again with another antibody overnight. Band intensities were measured by Bio-Profil Image analysis system (Vilber Lourmat, France) and were corrected forb-actin expression.
Statistical Analysis. Data are reported as mean6 S.E.M., and n represents the number of independent experiments for each in-dicated condition. Analyses were performed by SPSS 17.0 for Windows software (SPSS, Inc., Chicago, IL). Semiquantitative comparisons between the groups were calculated by Mann–Whitney U test. P , 0.05 (two-tailed) was considered statistically significant.
Results
Acute Stimulation ofa1-AR Transactivates EGFR in
A7R5 Cells. We recently had demonstrated that a1-AR
induced transactivation of EGFR contributes to VSMC con-traction in rat aorta (i.e., to immediate affect the a1-AR
response) (Ulu et al., 2010), so we first explored whether a similar transactivation occurs in serum-starved cultured A7R5 VSMCs, which express all three a1-ARs (data not
shown). Stimulation ofa1-AR by 10mM PE and EGFR by 1 nM
EGF induced phosphorylation of EGFR in A7R5 VSMCs. To examine the role of Ca21 entry in the transactivation, A7R5 cells were pretreated with prazosin (1mM) to fully block the increase in cellular Ca21 levels (data not shown). Whereas
prazosin fully blocked a1-AR–induced phosphorylation of
EGFR, it did not affect EGF-mediated EGFR phosphorylation (Supplemental Fig. 1A). Furthermore, the PE-induced in-crease in pEGFR was also antagonized by 10 mM AG1478 (Supplemental Fig. 1B).
All Three a1-AR Subtypes Transactivate EGFR in
CHO Cells. As acute stimulation ofa1-AR in A7R5 VSMCs
leads to EGFR transactivation, in the next step we addressed the question whether all three a1-AR subtypes can
trans-activate EGFR. To this end, CHO cells stably expressing a single subtype ofa1-AR and cotransfected with EGFR were
stimulated with 10mM PE or 1 nM EGF in the absence and presence of AG1478 (Fig. 1). The concentration of PE was selected as 10mM because a more pronounced phosphoryla-tion of EGFR was observed with this concentraphosphoryla-tion than with
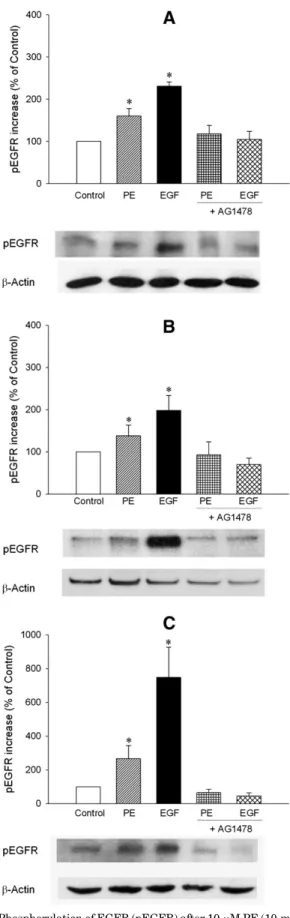
Fig. 1. Phosphorylation of EGFR (pEGFR) after 10mM PE (10-minute) or 1 nM EGF (10-minute) stimulation in the absence or presence of EGFR tyrosine kinase inhibitor AG1478 (30 minutes preincubation) in EGFR transfected CHO cells stably expressing (A)a1A-AR, (B)a1B-AR, or (C) a1D-AR. Representative Western blot detections of pEGFR andb-actin blots are shown. The pEGFR band intensities were corrected withb-actin and expressed as the percentage of control. Data are expressed as mean6 S.E.M. *P, 0.05 versus control.
at ASPET Journals on August 1, 2018
jpet.aspetjournals.org
1mM (Supplemental Fig. 2). In a1A-AR–expressing CHO cells
transfected with EGFR, both PE and EGF significantly increased the phosphorylation of EGFR (Fig. 1A; the per-centage increase in pEGFR was 160.06 18.1 and 231.3 6 9.6 for PE and EGF, respectively, n5 16–23). Likewise, PE and EGF significantly increased pEGFR ina1B-AR (Fig. 1B; the
percentage increase in pEGFR was 138.36 25.0 and 198.4 6 35.3 for PE and EGF, respectively, n5 4–15; P , 0.05 versus control for both) anda1D-AR (Fig. 1C; the percentage increase
in pEGFR was 266.56 77.1 and 748.2 6 178.8 for PE and EGF, respectively, n5 4–14, P , 0.05 versus control for both) expressing CHO cells. Increases in pEGFR mediated by all threea1-AR subtypes or EGF were antagonized by AG1478
(Fig. 1).
a1-AR–Mediated Transactivation of EGFR Is
Intra-cellular in CHO Cells. In the third step, we intended to identify the mechanism ofa1-AR–mediated transactivation of
EGFR. To pharmacologically determine whether transactiva-tion of EGFR bya1A-AR–expressing CHO cells was conveyed
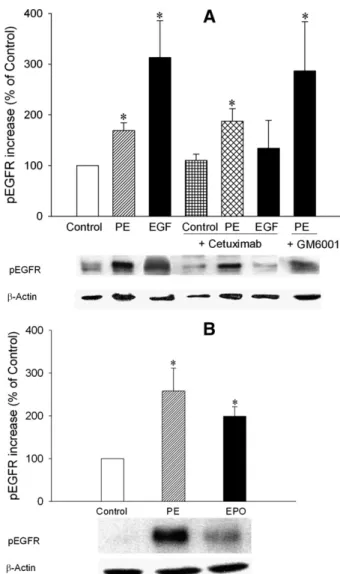
via an extracellular route, cells were pretreated with cetux-imab, precluding binding of EGF to the extracellular domain of EGFR (Graham et al., 2004) or GM6001, an MMP inhibitor limiting shedding of heparin-binding EGF-like growth factor (Hao et al., 2004). Cetuximab did not prevent the a1A
-AR–mediated increase in pEGFR (Fig. 2A; the percentage increase in pEGFR was 110.36 12.3 and 187.1 6 25.0 for control1 cetuximab and PE 1 cetuximab, respectively, n 5 8 for each condition), whereas the EGF-mediated increase in pEGFR was fully blocked (Fig. 2A; the percentage increase in pEGFR was 133.96 55.3 for EGF 1 cetuximab, n5 8; P , 0.05 versus EGF). Also, the inhibition of MMPs by GM6001 did not abrogate a1A-AR–induced
phosphory-lation of EGFR (Fig. 2A; the percentage increase in pEGFR was 286.5 6 97.5, n 5 7). The phosphorylation of EGFR appeared to be enhanced in the presence of GM6001, with a relatively higher S.E.M. In line with our observations, GM6001 was also reported previously as causing a slightly higher EGFR phosphorylation induced by insulin (Roztocil et al., 2008).
These experiments strongly suggest that transactivation of EGFR by a1A-AR depends on an intracellular route. To
conclusively demonstrate this, CHO cells were transfected with a chimeric EPOR-EGFR in which the extracellular domain of EGFR had been replaced by EPOR. In these cells,a1A-AR
stimulation by PE induced a substantial phosphorylation of the intracellular domain of the transiently expressed EPOR/ EGFR chimeric receptor (Fig. 2B; the percentage increase was 258.2 6 53.7, n 5 23; P , 0.05 versus control), as did EPO (Fig. 2B; the percentage increase was 198.86 22.7, n 5 23; P, 0.05 versus control). Collectively, these data demon-strate that transactivation of EGFR bya1A-AR depends on an
intracellular route.
We additionally examined this mechanism in a1D
-adrenoceptor-expressing CHO cells, asa1A-AR anda1D-AR are
predominantly found in VSMC (Piascik and Perez, 2001; Hein and Michel, 2007). Stimulation ofa1D-AR by PE in CHO cells
also induced phosphorylation of the intracellular domain of a transiently expressed EPOR/EGFR chimeric receptor (Supplemental Fig. 3A), which was abrogated by the EGFR kinase inhibitor AG1478. Thus, our data demonstrate that both a1A-AR anda1D-AR subtypes employ an intracellular route to
transactivate EGFR.
EGFR Transfection of CHO Cells Does Not Alter a1-AR–Mediated Intracellular Ca21. To substantiate the
functionality of a1-AR subtypes and to investigate the
contribution of EGFR transfection to a1-AR–mediated
in-tracellular Ca21responses, intracellular Ca21was measured in CHO cells. All three a1-AR subtypes induced an
intra-cellular Ca21response, of which thea1A-AR–induced increase
in fura-2 fluorescence was the highest compared witha1B-AR
and a1D-AR (Supplemental Fig. 4). Further, EGFR
trans-fection of CHO cells did not alter the intracellular Ca21 response mediated by all threea1-AR subtypes (Supplemental
Fig. 4). Thus, Ca21measurements indicate that all threea1
-AR subtypes are functional when stably transfected in CHO cells.
Fig. 2. Phosphorylation of EGFR (pEGFR) was investigated after 10mM PE (10-minute) or 1 nM EGF (10-minute) stimulation in the absence or presence of the anti-EGFR monoclonal antibody cetuximab (5mM, 2-hour preincubation) or broad spectrum MMP inhibitor GM6001 (10mM, 30-minute preincubation) in EGFR transfected CHO cells stably expressing a1A-AR (A). We also investigated pEGFR after 10mM PE (10-minute) or 100 IU×ml21EPO (10-minute) stimulation in EPOR-EGFR (extracellular domain of the EPOR fused to the transmembrane and intracellular domains of the EGFR) chimeric receptor transfected CHO cells stably expressinga1A-AR (B). Representative Western blot detections of pEGFR andb-actin blots are shown. The pEGFR band intensities were corrected withb-actin and expressed as the percentage of control. Data are expressed as mean6 S.E.M. *P , 0.05 versus control.
at ASPET Journals on August 1, 2018
jpet.aspetjournals.org
a1-AR–Mediated Transactivation of EGFR Involves
CaMKII, PI3K, and Src Activation, but Not the ERK1/2 and Akt Pathways. It is known that a1-AR stimulation
involves activation of Ca21/CaM, CaMKII, Src, PI3K, ERK1/2, and Akt; thus, in the next step, we sought to identify the roles and order of these signaling mediators in a1-AR–mediated
transactivation of EGFR. Hereto, CHO cells were stimulated witha1-AR or EGFR ligands in the absence and presence of
AG1478, GM60001, PD98059 (ERK1/2 inhibitor), PP2 (Src inhibitor), or LY294002 (PI3K inhibitor). We found thata1A
-AR stimulation by PE increased phosphorylation of CaMKII, which was blocked by Ca21/CaM inhibitor W7 (Fig. 3A; the percentage increase was 251.26 48.9 and 79.2 6 23.7 for PE and PE1 W7, respectively, n 5 17 for each condition; P , 0.05 for PE versus control). This finding substantiates the in-volvement of Ca21 as one of the major routes activated by a1-AR.
Likewise, PE induced increases in pSrc (Fig. 3B; the percentage increase was 436.26 205.1, n 5 12; P , 0.05 for PE versus control) and pPI3K (Fig. 3C, percentage increase was 238.46 75.4, n 5 7, P , 0.05 for PE versus control) were blocked by PP2 (Fig. 3B; the percentage increase was 42.66 8.0, n5 12) and LY294002 (Fig. 3C, the percentage increase was 93.7 6 50.6, n 5 7 independent experiments), respec-tively. Importantly,a1A-AR–mediated transactivation of
EGFR (Fig. 4; the percentage increase was 244.0 6 52.4, n 5 14) was completely blocked by preincubation with W7 (Fig. 4; the percentage increase was 117.86 40.6, n 5 17), PP2 (Fig. 4; the percentage increase was 156.56 36.3, n 5 12), LY294002 (Fig. 4; the percentage increase was 126.26 38.0, n5 7), and KN93 (Fig. 4; the percentage increase was 127.3 6 41.1, n5 14).
Similarly to thea1A-AR subtype, blockade of PI3K, Ca21/
CaM, and Src also attenuated the PE-induced phosphoryla-tion of EGFR in a1D-AR–expressing CHO cells (Supplemental
Fig. 3B), supporting the hypotheses that both a1-AR subtypes
employ a common intracellular pathway of EGFR transactivation. PE-induced increases in pSrc were blocked both by W7 (percentage increase was 84.5 6 27.5, n 5 15) and KN93 (percentage increase was 124.1 6 9.9, n 5 8), indicating involvement of Ca21/CaM and CaMKII upstream of Src (Fig. 5A). In addition, the PE-induced increase in pPI3K was blocked by W7 (percentage increase was 116.16 42.3, n 5 4), KN93 (percentage increase was 109.96 48.6, n 5 17), and PP2 (percentage increase was 95.36 23.0, n 5 6), indicating involvement of Src upstream of PI3K (Fig. 5B).
Stimulation of a1A-AR by PE also strongly increased
pERK1/2 (Fig. 6, B and C, the percentage increase was 3693.56 933.1, n 5 17; P , 0.05 for PE versus control), which was blocked both by LY294002 (Fig. 6C; percentage increase was 155.76 116.9, n 5 4) and PD98059 (Fig. 6C; percentage increase was 317.66 132.9, n 5 6), but not by AG1478 (Fig. 6, A and C; percentage increase was 3623.96 962.1, n 5 7; P , 0.05 for PE versus control). Moreover, the PE-induced increase in pEGFR was not antagonized by PD98059 (Fig. 6D; percentage increase was 239.36 75.5, n 5 7; P , 0.05 for
Fig. 3. Phosphorylations of calcium/calmodulin-dependent kinase II: (A) pCaMKII, (B) pSrc, and (C) pPI3K were investigated after 10mM PE (10-minute) stimulation in the absence or presence of calcium/calmodulin (Ca2+/CaM) inhibitor W7 (100mM, 30-minute preincubation), Src inhibitor PP2 (10mM, 30-minute preincubation), or PI3K inhibitor LY294002 (10 mM, 30-minute preincubation), respectively, in EGFR transfected CHO cells stably expressinga1A-AR. Representative Western blot detections of pCaMKII, pSrc, pPI3K, andb-actin blots are shown. The pCaMKII, pSrc,
and pPI3K band intensities were corrected withb-actin and expressed as the percentage of control. Data are expressed as mean6 S.E.M. *P , 0.05 versus control.
at ASPET Journals on August 1, 2018
jpet.aspetjournals.org
PE versus control), indicating that a1-AR–mediated
trans-activation of EGFR is independent of ERK1/2 trans-activation. These findings also demonstrate thata1A-AR–mediated
acti-vation of ERK1/2 is not through EGFR.
Likewise,a1A-AR stimulation by PE increased pAkt (Fig. 7,
A, B, and C; percentage increase was 223.96 19.3, n 5 14; P , 0.05 for PE versus control), which was blocked both by LY294002 (Fig. 7, B and C; percentage increase was 35.96 38.5, n5 3) and the Akt inhibitor MK2206 (Fig. 7, B and C; percentage increase was 65.4 6 31.3, n 5 14), but not by AG1478 (Fig. 7, A and C; percentage increase was 221.46 94.6, n5 4; P , 0.05 for PE versus control) or PD98059 (Fig. 7, A and C; percentage increase was 208.7 6 50.5, n 5 3). Moreover, the PE-induced increase in pEGFR was not an-tagonized by MK2206 (Fig. 7D; percentage increase was 254.96 53.0, n 5 11; P , 0.05 for PE versus control), indi-cating that a1-AR–mediated transactivation of EGFR is
in-dependent of Akt activation. These findings also demonstrate thata1A-AR–mediated activation of Akt is not through
trans-activation of EGFR.
Discussion
This study shows that all threea1-AR subtypes (a1A-AR,
a1B-AR, and a1D-AR) can transactivate the EGFR.
Trans-activation is dependent on an intracellular route rather than an extracellular route. First, EGFR transactivation is un-affected by cetuximab and the pan-MMP blocker GM6001, both acting extracellularly, whereas transactivation is blocked by AG1478, which acts intracellular. Second, a1A-AR and a1D-AR stimulation induced phosphorylation of
the intracellular domain of a transiently expressed EPOR/ EGFR chimeric receptor, which lacks the extracellular binding sites for EGF ligands. Collectively, these findings indicate that the transactivation of EGFR bya1A-AR anda1D-AR is
mediated by intracellular signaling. Further,a1A-AR stimulation
Fig. 4. Phosphorylation of EGFR (pEGFR) was investigated after 10mM PE (10-minute) stimulation in the absence or presence of calcium/ calmodulin (Ca2+/CaM) inhibitor W7 (100mM, 30-minute preincubation), Src inhibitor PP2 (10 mM, 30-minute preincubation), PI3K inhibitor LY294002 (10 mM, 30-minute preincubation), or calcium/calmodulin-dependent kinase II inhibitor KN93 (10mM, 30-minute preincubation), respectively, in EGFR transfected CHO cells stably expressinga1A-AR. Representative Western blot detections of pEGFR andb-actin blots are shown. The pEGFR band intensities were corrected with b-actin and expressed as the percentage of control. Data are expressed as mean6 S.E.M. *P, 0.05 versus control.
Fig. 5. Phosphorylation of Src (pSrc) was investigated after 10mM PE (10-minute) stimulation in the absence or presence of calcium/calmodulin (Ca2+/CaM) inhibitor W7 (100mM, 30-minute preincubation) or calcium/calmodulin-dependent kinase II inhibitor KN93 (10 mM, 30-minute preincubation) in EGFR transfected CHO cells stably expressinga1A-AR (A). Phosphorylation of pPI3K was also investigated after 10mM PE (10-minute) stimulation in the absence or presence of W7, KN93, or Src inhibitor PP2 (10 mM, 30-minute preincubation) in EGFR transfected CHO cells stably expressing a1A-AR (B). Representative Western blot detections of pSrc, pPI3K, andb-actin blots are shown. The pSrc and pPI3K band intensities were corrected with b-actin and expressed as the percentage of control. Data are expressed as mean6 S.E.M. *P , 0.05 versus control.
at ASPET Journals on August 1, 2018
jpet.aspetjournals.org
enhanced phosphorylation of ERK1/2 and Akt, and both were unaffected by the EGFR kinase inhibitor AG1478, indicating that ERK1/2 and Akt phosphorylation is independent of EGFR trans-activation. Accordingly, inhibitors of ERK1/2 or Akt did not influence the a1A-AR–mediated EGFR transactivation.
Im-portantly, inhibition of CaMKII, Src, and PI3K blocked both a1A-AR– and a1D-AR–mediated EGFR transactivation.
Col-lectively, these results demonstrate an intracellular EGFR transactivation route involving the phosphorylation of CaM-KII, Src, and PI3K, but not of ERK1/2 and Akt pathways in CHO cells.
Several studies have been performed to identify the exact mechanism of EGFR transactivation mediated by GPCRs. Proteolytic metalloprotease-dependent shedding of EGFR ligands has been proposed as a key regulator of EGFR sig-naling for heart development and angiogenesis (Dong et al., 1999; Iwamoto et al., 2003; Kurohara et al., 2004; Zhou et al., 2004). Moreover, it has been postulated that cross-talk between GPCRs (including a1-ARs) and EGFR is mediated
via MMP-dependent shedding of EGFR ligands (Prenzel et al., 1999; Asakura et al., 2002; Zhang et al., 2004). In our recent and present studies, we sought to identify the mechanism of a1-AR–dependent EGFR transactivation. In this study,
neither EGFR monoclonal antibody (cetuximab) nor broad
spectrum MMP inhibitor (GM60001) antagonized PE-mediated EGFR phosphorylation. Furthermore, we took a novel approach to conclusively determine whether an intra-cellular or extraintra-cellular signaling route is involved in the transactivation of EGFR. In these experiments, PE induced phosphorylation of the intracellular part of EPOR-EGFR chimeric receptor, which lacks binding sites for EGFR ligands. These results strongly support that the transactivation of EGFR by a1-AR is mediated by intracellular signaling
molecules.
On the one hand, acute stimulation ofa1-ARs by
catechol-amines enhances vascular tone by the activation of direct signaling pathways involving Gqand PI3K and elevation of
intracellular Ca21. On the other hand, the present study and
earlier studies indicate that acute and prolonged stimulation ofa1-ARs also influences the cellular function by an indirect
route consisting of transactivation of EGFR (Asakura et al., 2002; Zhang et al., 2004; Ulu et al., 2010; Li et al., 2011). It is obvious that based on the localization ofa1-ARs this signaling
has physiologic and pathologic outcomes, particularly in CV diseases. Of the three differenta1-AR subtypes,a1A-AR and
a1B-AR are mainly localized in the myocardium, anda1A-AR
and a1D-AR are predominant in VSMC (Piascik and Perez,
2001; Hein and Michel, 2007). As this study shows, all three
Fig. 6. Phosphorylation of extracellular signal regulated kinases (pERK1/2) was investigated after 10mM PE (10-minute) or 1 nM epidermal growth factor (EGF, 10-minute) stimulations in the absence or presence of EGFR tyrosine kinase inhibitor AG1478 (30-minute preincubation) (A), phosphatidylinositol 3-kinase inhibitor LY294002 (10mM, 30-minute preincubation), or ERK1/2 inhibitor PD98059 (10 mM, 30-minute preincubation) (B) in EGFR transfected CHO cells stably expressinga1A-AR. Phosphorylation of pEGFR was also investigated after 10mM PE (10-minute) stimulation in the absence or presence of PD98059 in EGFR transfected CHO cells stably expressinga1A-AR (D). Representative Western blot detections of pERK1/2, pEGFR, andb-actin blots are shown. The pERK1/2 and pEGFR band intensities were corrected with b-actin and expressed as the percentage of control (C and D). Data are expressed as mean6 S.E.M. *P , 0.05 versus control.
at ASPET Journals on August 1, 2018
jpet.aspetjournals.org
a1-AR subtypes are able to transactivate EGFR, so EGFR
signaling seems be an alternative target in CV diseases of different origin (Ulu et al., 2013).
We observed that a1A-AR stimulation activated Akt and
ERK1/2 via an EGFR independent route, as blockade of EGFR phosphorylation by AG1478 did not antagonize a1A-AR–
mediated activation of Akt or ERK1/2. Conversely, inhibitors of Akt or ERK1/2 did not influence the phosphorylation of EGFR after PE stimulation, which precludes the involve-ment of Akt or ERK1/2 ina1A-AR–mediated transactivation.
All three a1-AR subtypes couple to Gqand result in
acti-vation of phospholipase C with an increase in inositol triphosphate and subsequent mobilization of intracellular Ca21, which leads to activation of the calcium/calmodulin (Ca21/CaM) pathway (Wu et al., 1992; Bylund et al., 1994;
Della Rocca et al., 1997; Koshimizu et al., 2003). The Ca21/CaM pathway directly and indirectly influences EGFR and has a regulatory function in EGFR signaling (Murasawa et al., 1998; Aifa et al., 2002; Li and Villalobo, 2002; Sanchez-Gonzalez et al., 2010). Other important signaling molecules activated by
a1-AR are the Src (Han et al., 2008) and PI3K, which are also
implicated in EGFR transactivation.
In this study, we found thata1A-AR stimulation activated
CaMKII, indicating the activation of Ca21/CaM pathway.
Moreover, Ca21/CaM and CaMKII inhibitors (W7 and KN93, respectively) not only blockeda1A-AR– and a1D-AR–mediated
EGFR transactivation but also the phosphorylation of Src and PI3K. Consequently, CaMKII activation seems to represent the most upstream event in transactivation of EGFR. Stim-ulation ofa1A-AR also increased the PI3K and Src activity,
and inhibitors of Src and PI3K (PP2 and LY294002, re-spectively) antagonizeda1A-AR– and a1D-AR–mediated EGFR
phosphorylation. Furthermore, as the PE-induced activation of PI3K was also blocked by PP2, Src activation seems to be upstream of PI3K.
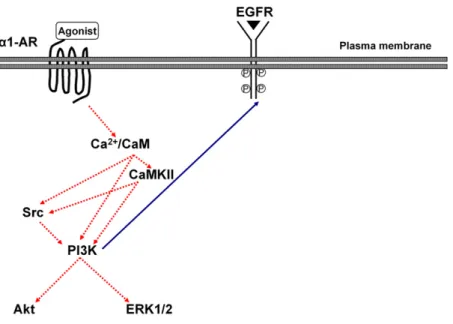
Taken together, the cascade of a1A-AR- and a1D-AR–
mediated transactivation of EGFR is triggered by Ca21 influx and subsequent Ca21/CaM and CaMKII, which triggers signaling through Src and PI3K, as depicted in Fig. 8. This activation cascade is consistent with previous
Fig. 7. Phosphorylation of serine-threonine kinases (pAkt) was investigated after 10mM PE (10-minute) stimulation in the absence or presence of EGFR tyrosine kinase inhibitor AG1478 (30-minute preincubation), extracellular signal regulated kinases inhibitor PD98059 (10 mM, 30-minute preincubation) (A), phosphatidylinositol 3-kinase inhibitor LY294002 (10mM, 30-minute preincubation), or Akt inhibitor MK2206 (1 mM, 30-minute preincubation) (B) in EGFR transfected CHO cells stably expressinga1A-AR. Phosphorylation of pEGFR was also investigated after 10 mM PE (10-minute) stimulation in the absence or presence of MK2206 in EGFR transfected CHO cells stably expressinga1A-AR (D). Representative Western blot detections of pAkt, pEGFR, andb-actin blots are shown. The pAkt and pEGFR band intensities were corrected with b-actin and expressed as the percentage of control (C and D). Data are expressed as mean6 S.E.M. *P , 0.05 versus control.
at ASPET Journals on August 1, 2018
jpet.aspetjournals.org
reports (Della Rocca et al., 1997; Ballou et al., 2000; Gentili et al., 2002; Kubo et al., 2005). These results extend our findings for an intracellular route involving a Ca21 /CaM-CaMKII pathway, PI3K, and Src, but not Akt or ERK1/2 for a1A-AR–mediated transactivation of EGFR. In line with our
results, it was previously reported that a1-AR–mediated
growth of adventitial cells activation of the downstream ERK1/2 pathway does not mediate the transactivation of EGFR (Zhang et al., 2004).
The pathway identified in our present study may be of significant physiologic and pathophysiologic importance, and interruption of EGFR transactivation by targeting one of the dissected key components may provide a novel approach in the treatment of CV pathologies. The main limitation of our study is that we investigated the mechanism of a1-AR–
mediated EGFR transactivation in an isolated CHO cell culture model system. Therefore, the mechanism of this transactivation both in VSCM and under in vivo conditions still requires confirmation. Furthermore, our data do not provide conclusive evidence on the role of a1-AR–mediated
EGFR transactivation in vascular hypertrophy and VSMC proliferation. Further research using specific assays in cell culture and animal models will allow testing the role of the proposed mechanism in CV hypertrophy and proliferation.
Our study demonstrates that all a1-AR subtypes can
transactivate EGFR, which is dependent on an intracellular route involving CaMKII, PI3K, and Src activation but is independent from ERK1/2 and Akt in CHO cells. Moreover, we demonstrate that a1A-AR–dependent activation of Akt
or ERK1/2 is independent of EGFR transactivation. There-fore, our study not only establishes a1A-AR– and a1D-AR–
dependent transactivation of EGFR to be governed by the intracellular route, but also extends the evidence that Akt and ERK1/2 need not necessarily require cross-talk of GPCRs with receptor tyrosine kinases.
Acknowledgments
The authors thank Prof. Dr. Mehmet Ugur (Department of Biophysics, Faculty of Medicine, University of Ankara, Ankara, Turkey) for expert technical assistance on the intracellular calcium measurements.
Authorship Contributions
Participated in research design: Ulu, Henning, Gurdal.
Conducted experiments: Ulu, Guner, Zoto, Duman-Dalkilic, Duin, Gurdal.
Contributed new reagents or analytic tools: Ulu, Henning, Guner, Duman-Dalkilic, Gurdal.
Performed data analysis: Ulu, Henning, Guner, Zoto, Duman-Dalkilic, Duin, Gurdal.
Wrote or contributed to the writing of the manuscript: Ulu, Henning, Gurdal.
References
Aifa S, Johansen K, Nilsson UK, Liedberg B, Lundström I, and Svensson SP (2002) Interactions between the juxtamembrane domain of the EGFR and calmodulin measured by surface plasmon resonance. Cell Signal 14:1005–1013.
Asakura M, Kitakaze M, Takashima S, Liao Y, Ishikura F, Yoshinaka T, Ohmoto H, Node K, Yoshino K, and Ishiguro H, et al. (2002) Cardiac hypertrophy is inhibited by antagonism of ADAM12 processing of HB-EGF: metalloproteinase inhibitors as a new therapy. Nat Med 8:35–40.
Ballou LM, Cross ME, Huang S, McReynolds EM, Zhang BX, and Lin RZ (2000) Differ-ential regulation of the phosphatidylinositol 3-kinase/Akt and p70 S6 kinase pathways by the alpha(1A)-adrenergic receptor in rat-1 fibroblasts. J Biol Chem 275:4803–4809. Barki-Harrington L, Perrino C, and Rockman HA (2004) Network integration of the
adrenergic system in cardiac hypertrophy. Cardiovasc Res 63:391–402. Bylund DB, Eikenberg DC, Hieble JP, Langer SZ, Lefkowitz RJ, Minneman KP,
Molinoff PB, Ruffolo RR, Jr, and Trendelenburg U (1994) International Union of Pharmacology nomenclature of adrenoceptors. Pharmacol Rev 46:121–136. Chen L, Xin X, Eckhart AD, Yang N, and Faber JE (1995) Regulation of vascular
smooth muscle growth by alpha 1-adrenoreceptor subtypes in vitro and in situ. J Biol Chem 270:30980–30988.
Cipolletta E, Monaco S, Maione AS, Vitiello L, Campiglia P, Pastore L, Franchini C, Novellino E, Limongelli V, and Bayer KU, et al. (2010) Calmodulin-dependent kinase II mediates vascular smooth muscle cell proliferation and is potentiated by extracellular signal regulated kinase. Endocrinology 151:2747–2759.
Della Rocca GJ, van Biesen T, Daaka Y, Luttrell DK, Luttrell LM, and Lefkowitz RJ (1997) Ras-dependent mitogen-activated protein kinase activation by G protein-coupled receptors. Convergence of Gi- and Gq-mediated pathways on calcium/ calmodulin, Pyk2, and Src kinase. J Biol Chem 272:19125–19132.
Dong J, Opresko LK, Dempsey PJ, Lauffenburger DA, Coffey RJ, and Wiley HS (1999) Metalloprotease-mediated ligand release regulates autocrine signaling through the epidermal growth factor receptor. Proc Natl Acad Sci USA 96: 6235–6240.
Engelhardt S (2007) Alternative signaling: cardiomyocyte beta1-adrenergic receptors signal through EGFRs. J Clin Invest 117:2396–2398.
Gentili C, Morelli S, and Russo De Boland A (2002) Involvement of PI3-kinase and its association with c-Src in PTH-stimulated rat enterocytes. J Cell Biochem 86:773–783. Graham J, Muhsin M, and Kirkpatrick P (2004) Cetuximab. Nat Rev Drug Discov 3:
549–550.
Haba M, Hatakeyama N, Kinoshita H, Teramae H, Azma T, Hatano Y, and Matsuda N (2010) The modulation of vascular ATP-sensitive K1 channel function via the phosphatidylinositol 3-kinase-Akt pathway activated by phenylephrine. J Phar-macol Exp Ther 334:673–678.
Han C, Bowen WC, Michalopoulos GK, and Wu T (2008) Alpha-1 adrenergic receptor transactivates signal transducer and activator of transcription-3 (Stat3) through activation of Src and epidermal growth factor receptor (EGFR) in hepatocytes. J Cell Physiol 216:486–497.
Fig. 8. Model ofa1-AR–mediated transactivation of EGFR in CHO cells. Activation of thea1-AR leads to EGFR trans-activation via an intracellular mechanism involving calcium/ calmodulin (Ca2+/CaM), calcium/calmodulin-dependent ki-nase II (CaMKII), phosphatidylinositol 3-kiki-nase (PI3K), and Src activation but not the extracellular signal regulated kinases (ERK1/2) and serine-threonine kinases (Akt).
at ASPET Journals on August 1, 2018
jpet.aspetjournals.org
Hao L, Du M, Lopez-Campistrous A, and Fernandez-Patron C (2004) Agonist-induced activation of matrix metalloproteinase-7 promotes vasoconstriction through the epidermal growth factor-receptor pathway. Circ Res 94:68–76.
Hein P and Michel MC (2007) Signal transduction and regulation: are all alpha1-adrenergic receptor subtypes created equal? Biochem Pharmacol 73:1097–1106. Illario M, Cavallo AL, Bayer KU, Di Matola T, Fenzi G, Rossi G, and Vitale M (2003)
Calcium/calmodulin-dependent protein kinase II binds to Raf-1 and modulates integrin-stimulated ERK activation. J Biol Chem 278:45101–45108.
Iwamoto R, Yamazaki S, Asakura M, Takashima S, Hasuwa H, Miyado K, Adachi S, Kitakaze M, Hashimoto K, and Raab G, et al. (2003) Heparin-binding EGF-like growth factor and ErbB signaling is essential for heart function. Proc Natl Acad Sci USA 100:3221–3226.
Koshimizu TA, Tanoue A, Hirasawa A, Yamauchi J, and Tsujimoto G (2003) Recent advances ina1-adrenoceptor pharmacology. Pharmacol Ther 98:235–244. Kubo H, Hazeki K, Takasuga S, and Hazeki O (2005) Specific role for p85/p110b in
GTP-binding-protein-mediated activation of Akt. Biochem J 392:607–614. Kurohara K, Komatsu K, Kurisaki T, Masuda A, Irie N, Asano M, Sudo K,
Nabe-shima Y, Iwakura Y, and Sehara-Fujisawa A (2004) Essential roles of Meltrin beta (ADAM19) in heart development. Dev Biol 267:14–28.
Li H and Villalobo A (2002) Evidence for the direct interaction between calmodulin and the human epidermal growth factor receptor. Biochem J 362:499–505. Li Y, Zhang H, Liao W, Song Y, Ma X, Chen C, Lu Z, Li Z, and Zhang Y (2011)
Transactivated EGFR mediatesa1-AR-induced STAT3 activation and cardiac
hy-pertrophy. Am J Physiol Heart Circ Physiol 301:H1941–H1951.
Luttrell LM (2005) Composition and function of g protein-coupled receptor signal-somes controlling mitogen-activated protein kinase activity. J Mol Neurosci 26: 253–264.
Murasawa S, Mori Y, Nozawa Y, Gotoh N, Shibuya M, Masaki H, Maruyama K, Tsutsumi Y, Moriguchi Y, and Shibazaki Y, et al. (1998) Angiotensin II type 1 receptor-induced extracellular signal-regulated protein kinase activation is medi-ated by Ca21/calmodulin-dependent transactivation of epidermal growth factor receptor. Circ Res 82:1338–1348.
Piascik MT and Perez DM (2001)a1-adrenergic receptors: new insights and direc-tions. J Pharmacol Exp Ther 298:403–410.
Prenzel N, Zwick E, Daub H, Leserer M, Abraham R, Wallasch C, and Ullrich A (1999) EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature 402:884–888.
Roztocil E, Nicholl SM, and Davies MG (2008) Insulin-induced epidermal growth factor activation in vascular smooth muscle cells is ADAM-dependent. Surgery 144: 245–251.
Sánchez-González P, Jellali K, and Villalobo A (2010) Calmodulin-mediated regula-tion of the epidermal growth factor receptor. FEBS J 277:327–342.
Ulu N, Gurdal H, Landheer SW, Duin M, Guc MO, Buikema H, and Henning RH (2010)a1-Adrenoceptor-mediated contraction of rat aorta is partly mediated via
transactivation of the epidermal growth factor receptor. Br J Pharmacol 161: 1301–1310.
Ulu N, Mulder GM, Vavrinec P, Landheer SW, Duman-Dalkilic B, Gurdal H, Goris M, Duin M, van Dokkum RP, and Buikema H, et al. (2013) Epidermal growth factor receptor inhibitor PKI-166 governs cardiovascular protection without beneficial effects on the kidney in hypertensive 5/6 nephrectomized rats. J Pharmacol Exp Ther 345:393–403.
Wu D, Katz A, Lee CH, and Simon MI (1992) Activation of phospholipase C by alpha 1-adrenergic receptors is mediated by the alpha subunits of Gq family. J Biol Chem 267:25798–25802.
Xiao L, Pimental DR, Amin JK, Singh K, Sawyer DB, and Colucci WS (2001) MEK1/ 2-ERK1/2 mediatesa1-adrenergic receptor-stimulated hypertrophy in adult rat
ventricular myocytes. J Mol Cell Cardiol 33:779–787.
Zhang H, Chalothorn D, Jackson LF, Lee DC, and Faber JE (2004) Transactivation of epidermal growth factor receptor mediates catecholamine-induced growth of vas-cular smooth muscle. Circ Res 95:989–997.
Zhou HM, Weskamp G, Chesneau V, Sahin U, Vortkamp A, Horiuchi K, Chiusaroli R, Hahn R, Wilkes D, and Fisher P, et al. (2004) Essential role for ADAM19 in car-diovascular morphogenesis. Mol Cell Biol 24:96–104.
Zhu HJ, Iaria J, Orchard S, Walker F, and Burgess AW (2003) Epidermal growth factor receptor: association of extracellular domain negatively regulates in-tracellular kinase activation in the absence of ligand. Growth Factors 21:15–30.
Address correspondence to: Dr. Hakan Gurdal, Professor of Pharmacology, Department of Medical Pharmacology, Faculty of Medicine, University of Ankara, Ankara Universitesi Tip Fakultesi, Tibbi Farmakoloji Anabilim Dali, Morfoloji Binasi Kat: 1 Sihhiye, 06100, Ankara, Turkey. E-mail:gurdal@ medicine.ankara.edu.tr
at ASPET Journals on August 1, 2018
jpet.aspetjournals.org