Contents lists available atScienceDirect
European Journal of Pharmaceutical Sciences
journal homepage:www.elsevier.com/locate/ejpsMulti-targeted anti-leukemic drug design with the incorporation of silicon
into Nelarabine: How silicon increases bioactivity
Esma Eryilmaz
Selcuk University, Faculty of Technology, Department of Biomedical Engineering, 42031 Konya, Turkey
A R T I C L E I N F O Keywords: Pharmacokinetic ADME In silico Anti-leukemia Silicon Drug-likeness A B S T R A C T
Acute Lymphoblastic Leukemia (ALL) represents 30% of all childhood cancers and children younger than 5 years old have the highest risk for developing ALL. Existing ALL drugs do not respond in approximately 20% of treatment. Therefore, drug development studies against ALL must be continued with either developing existing drugs or discovering new ones. In this study, we evaluated the U.S Food and Drug Administration (FDA) ap-proved ALL drugs according to their physicochemical and pharmaceutical properties, and Nelarabine was found to have the highest bioactivity score. Using the key strategy of bioisosterism commonly accepted by medicinal chemists, we investigated in silico ADME properties, drug-likeness, and biological activity of new designed twenty-four compounds including Nelarabine. The results were evaluated in terms of two classifications: broad spectrum biological activity and filtering of five different drug likeness criteria of the literature including Lipinski's rule of five. We interestingly observed that silicon incorporated compounds exhibited better perfor-mance on both criteria by targeting broader spectrum of drug receptors including G-protein coupled receptor (GPCR), ion channel modulator, kinase inhibitor, protease and enzyme inhibitor and by satisfying all of five different drug-likeness criteria reported in the literature. Design compound C19 appeared as a potential drug candidate for further pharmacological research.
1. Introduction
Leukemias are cancers that start mostly in early form of white blood cells and generally in other types of blood cells.
According to the American Cancer Society, Acute Lymphoblastic Leukemia (ALL) also called acute lymphocytic leukemia is a severe type of leukemia progressing quickly and can be fatal within a few months if not treated. ALL occurs mostly in children and represents 30% of all childhood cancers. Children younger than 5 years old have the highest risk for developing ALL. Although the existing ALL drugs treat 80% of the disease, the causes of the remaining part are still unknown. And yet, a significant amount of patients was found to have either resistance or sensitivity to cancer drugs (Holleman et al., 2004; Gottesman, 2002; Holohan et al., 2013). Holleman and co-worker's study was performed with leukemia cells taken from 173 patients for the purpose of testing for sensitivity and resistance to the common ALL drugs prednisole, vincristine, asparaginase, and daunorubicin. The results showed that about 50% of patients exhibited either sensitivity or resistant to the given drugs (Holleman et al., 2004). To overcome the resistance and sensitivity problem of available drugs, to decrease adverse effects, and to improve efficacy, more studies on the development of the existing antileukemic drugs or designing new ones are obligatory.
In target-based drug design approach, investigating ligands with maximum selectivity on a specific target has been found a common place for many researchers. However, a number of complex diseases such as cancer is resulted from multi misfunctional entities requiring more than one specific targeted medicine. For that, multiple targeted agents have been shown to have improved therapeutic efficacy. Fu's review on multi-targeted anticancer agents would be worth to mention here (Fu et al., 2017). The distribution of first-in-class drugs according to target family showed that G-protein coupled receptors (GPCR), ki-nases, proteases, and ion channels are the major drug targets (Hopkins and Groom, 2002). Based on that, we, in this study, investigated the relative biological activity of new design antileukemic compounds in terms of multi-targeted manner. We tried to obtain a better design than the approved drugs exhibiting the broadest activity on the most im-portant drug targets such as GCPR, kinase, nuclear receptors, ion channel modulators, protease, and enzyme inhibitors.
New molecule design and drug discovery research is largely de-pendent upon bioinformatics, since it is basically dealing with a number of small molecular database, molecular physico-chemical, pharmoco-logical properties and molecular structure-function relationship. So called with Computer-Aided Drug Design (CADD), one can design a new therapeutic agent using molecular modeling, theoretically predict its
https://doi.org/10.1016/j.ejps.2019.04.008
Received 31 January 2019; Received in revised form 19 March 2019; Accepted 4 April 2019
E-mail address:[email protected].
Available online 25 April 2019
0928-0987/ © 2019 Elsevier B.V. All rights reserved.
molecular properties using quantum chemical calculations, improve its desired properties, and diminish its possible side effects using bioin-formatics prediction softwares. In this study, we used Molinspiration, SwissADME, and Avogadro softwares as bioinformatics and che-minformatics resources.
A common way of investigating new therapeutic agents is the re-placement of bioisesteric members into known drugs. Bioisosteres are atoms or groups of molecules that have similar physicokinetic proper-ties and produce similar biological effect. Bioisosteres are widely clas-sified as classical and non-classical ones. Monovalent groups of classical bioisosteres are ordered as follows: eOH, eNH2, eCH3, eOR, eF, eCl, eBr, eI, eSH, -PH2, eSi3. In this study, using the concepts of bioisos-terism, in silico ADME properties, and ‘drug-likeness’ filtering, we de-signed new molecular compounds and evaluated them in terms of their bioactivity performance and druggability potential by computational modeling approach and bio & cheminformatics prediction softwares. 2. Materials and methods
The list of the U.S Food and Drug Administration (FDA) approved drugs for ALL were obtained from the official website of the National Cancer Institute, the National Institute of Health of USA (NIH),https:// www.cancer.gov/. From the list, we, exclusively, selected small mole-cule drugs rather than drug combinations or biotechnological ones. 2.1. Ligand molecule design and preparation
SMILES codes of the FDA approved drugs were obtained from the unique bioinformatics and cheminformatics resource, DrugBank 5.1.1 https://www.drugbank.ca/ (Wishart et al., 2017). FDA approval his-tory of the marketed drugs were found in the database ofwww.drugs. com. The list of antileukemic small molecule drugs and their DrugBank identification numbers are as follows: Clofarabine DB00631, Cyclo-phosphamide DB00531, Cytarabine DB00987, Dasatinib DB01254, Daunorubicin DB00694, Doxorubicin DB00997, Lapatinib DB01259, Mercaptopurine DB01033, Methotrexate DB00563, Nelarabine DB01280, Prednisone DB00635, Ponatinib DB08901, Venetoclax DB11581, Vincristine DB00541.
2D structures of the anti-leukemic drugs shown in Fig. 1 were produced from their SMILES notations using Molinspiration and ver-ified by SwissADME softwares.
2.2. Calculation of pharmacokinetic parameters
SMILES notations of the drug molecules were fed into the Molinspiration cheminformatics software to calculate molecular phy-sicochemical properties and to predict bioactivity scores (https://www. molinspiration.com/). Molecular physicochemical properties of the listed drugs such as partition coefficient (Log P), topological polar surface area (TPSA), number of hydrogen bond donor and hydrogen bond acceptors, the number of rotatable bonds, molecular volume and molecular weight, number of heavy atoms, number of drug-likeness violations were calculated and presented inTable 1.
2.3. Prediction of bioactivity
Each drug molecule was evaluated in terms of its biological activity for the most important drug targets such as G-protein coupled receptors (GPCR), kinase inhibitors, nuclear receptors, ion channel modulators, protease and enzyme inhibitors by using Molinspiration. The virtual screening results of the software are represented as color coded. Properties shown in dark green indicate a good bioactivity behaviour of the lead molecule for the listed drug targets. For the detailed drug likeness properties, free online cheminformatics tool SwissADME de-veloped by Swiss Institute of Bioinformatics was used to calculate physicochemical descriptors and to predict ADME parameters of both
approved drug molecules and the design compounds. 2.4. Evaluation of “drug-likeness” score and criteria
For computational drug-likeness evaluation of a new designed compound, the molecular physicochemical properties such as lipophi-licity, polar surface area, number of hydrogen bond donor and receptor, molecular weight and volume are used as descriptors of the molecule. Using those properties, Lipinski (Lipinski, 2001), Ghose (Ghose et al., 1999), Veber (Veber et al., 2002), Egan (Egan et al., 2000), and Muegge (Muegge et al., 2001) were published their own drug-likeness criteria summarized in Table 2, and the Lipinski's rule of five is commonly applied. For this study, we examined the design compounds in terms of two criteria; one is “drug-likeness” scores and the other one is biological activity performance in terms of six most important drug targets listed above. Bioactivity scores of the marketed FDA drugs and the design molecules for the listed drug targets, GPCR, kinase inhibitor, ion channel modulator, nuclear receptor, protease inhibitor, and enzyme inhibitor, were predicted by Molinspiration and tabulated inTable 3 andTable 4, respectively. A positive bioactivity score equal or higher than 0.5 is considered as a sign of good biological activity of the mo-lecule of interest by colouring with dark green and moderate activity is colored by light green for scores equal or higher than 0.2.
2.5. Bioisosteric replacement
Bioisosterism is a widely used concept in drug discovery process for the investigation of a lead molecule having better pharmacokinetic properties. In this study, we are interested in designing novel bioactive molecules for ALL treatment with less side effect, decreased toxicity and more efficient impact on the disease. The replacement of the mono-valent groups of classical bioisosteres, −OH, − NH2, − CH3, − OR, − F, − Cl, − Br, − I, − SH, − PH2, − Si3 on the lead molecule Nelarrabine was investigated in terms of biological activity and drug-likeness properties. The position of bioisosteric replacement of NH2and CH3groups of the marketed antileukemic drug molecule Nelarabine, the design molecules C1-C23, and their corresponding bioactivity score were provided inTable 4.
3. Results
3.1. In silico physicochemical properties
Some basic molecular structural and physicochemical properties such as Log P, molecular weight, number of rotatable bonds, number of hydrogen bond donor and hydrogen bond acceptor are used as key indicators of a molecule for being a good oral bioavailable drug. Log P (P is the calculated partition coefficient of the molecule in octanol/ water system) is recently often used as an important descriptor of drug candidate in rational drug design studies. It is accepted as an indicator of molecular hydrophobicity or lipophilicity. It is effective in many contents related to drug discovery including solubility (Log D), toxicity such as phospholipidosis (Hanumegowda et al., 2010), cytochrome inhibition, and even in the binding affinity to target protein. For more detail of lipophilicity on drug discovery, Waring's review is worth to mention here (Waring, 2010). Positive value of Log P represents hy-drophobic or lipophilic nature of molecule, while negative Log P re-presents more hydrophilic nature. All ‘drug-likeness models’ reported in the literature and listed inTable 2suggested Log P value to be equal or < 5. By examining multi-targeted activity of all marketed drugs, Nelarabine was chosen as a lead molecule which was explained in detail insubsection 3.3. Our lead molecule Nelarabine showed a negative Log P value of −1.28 indicating the hydrophilic nature, while the silicon incorporated design compound C19 resulted in 3.91 of Log P indicating a comparatively much higher hydrophobic or lipophilic nature (Table 5). For example, silicon incorporated unnatural amino acid
silaproline was experimentally shown to be active in contrast to proline even in the absence of protease inhibitor. That was interpreted to the higher stability of the compound in vivo. The reason of the improved stability was correlated to 14 times greater cLogP value of Fmoc-sila-proline than that of Fmoc-Fmoc-sila-proline (Cavelier et al., 2002). Lipophilicity will be discussed more in detail insubsection 3.5.2.
Molecular weight and volume which are related to size of the mo-lecule are effected in solubility and diffusion of the momo-lecule trough the lipid bilayer membrane. For that reason, the range of molecular weight is preferred to be between 160 and 480 g/mol and < 500 g/mol ac-cording to Ghose's (Ghose et al., 1999) and Lipinski's (Lipinski, 2001) criteria, respectively. All of four promising design compounds and Nelarabine fall into this range. With regard to polarity, TPSA is con-sidered to be in a certain range for a good oral bioavailable drug, since polar molecules dissociate better than non-polar molecules to some level which was limited to equal or < 150 Å2 according to Muegge's criteria (Table 2). From our design compounds (C1-C23), the four promising ones were listed in Table 5 according to their biological
activity across drug target. From the compounds in Table 5, C21 showed higher TPSA value than the upper limit of Muegge's, Veber's, and Eagan's criteria summarized in Table 2. C18 and Nelarabine showed a very close value, 148.78 Å2, of the upper limit of Lipinski's rule, 150 Å2. However, silicon incorporated compounds C19 (122.76 Å2) and C20 (122.76 Å2) showed a better profile for TPSA value which satisfied Muegge's (150), Veber's (140) and Egans's (131.6) cri-teria.
Hydrogen bonding capacity of a drug with the number of donors and acceptors is another important factor for a good oral bioavailable drug candidate as an indicator of good membrane permeability (Refsgaard et al., 2005). Both of Lipinski's and Muegge's criteria sum-marized inTable 2suggested that having > 5 hydrogen bond donors and 10 hydrogen bong acceptors could be the cause of poor cell membrane permeation and absorption. From the compound series of C1 to C23 described inTable 4and from the chosen four promising com-pounds (C18–C21) inTable 5, C21 again exceeded both of the criteria with 11 HBA. The rest three compounds C18, C19, C20 and Nelarabine satisfied the hydrogen binding rules.
Restricted number of rotatable bond is another criteria for oral bioavailability. The more rotatable bond gives larger conformational flexibility to the molecule which is not preferential for a stable drug-target or drug-channel interaction. This criteria found a place in both Veber's and Ghose's filter for being equal or < 10 and 15, respectively. All of four design compounds (C18–C21) and marketed drug Nelarabine showed an appropriate number of rotatable bonds. Higher number of rotatable bond ofC19 provides for a little more conformational flex-ibility than Nelarabine that could make the finding the best pose easier to Nelarabine during the binding process.
3.2. Evaluation of druglikeness criteria
Although “Lipinski's rule of five” is widely applied for druglikeness evaluation, we included and discussed the other filtering criteria pro-vided in the literature. We evaluated four design compounds chosen from C1 to C23 series showing the highest bioactivity, in terms of Chose's, Veber's, Egan's, Muegge's, and Lpinski's drug-likeness rules using SwissADME tool. The results provided inTable 5showed that the new design C19 is a more potent drug candidate satisfying all of five criteria, although Nelarabine obeyed only two of them. The compound C20 showed also a good profile resulting in only one violation in terms of five druglikeness criteria.
Fig. 1. Chemical structures and names of the existing FDA approved anti-leukemic drugs. SMILES codes of the drug molecules are provided in Supplementary
MaterialTable 2.
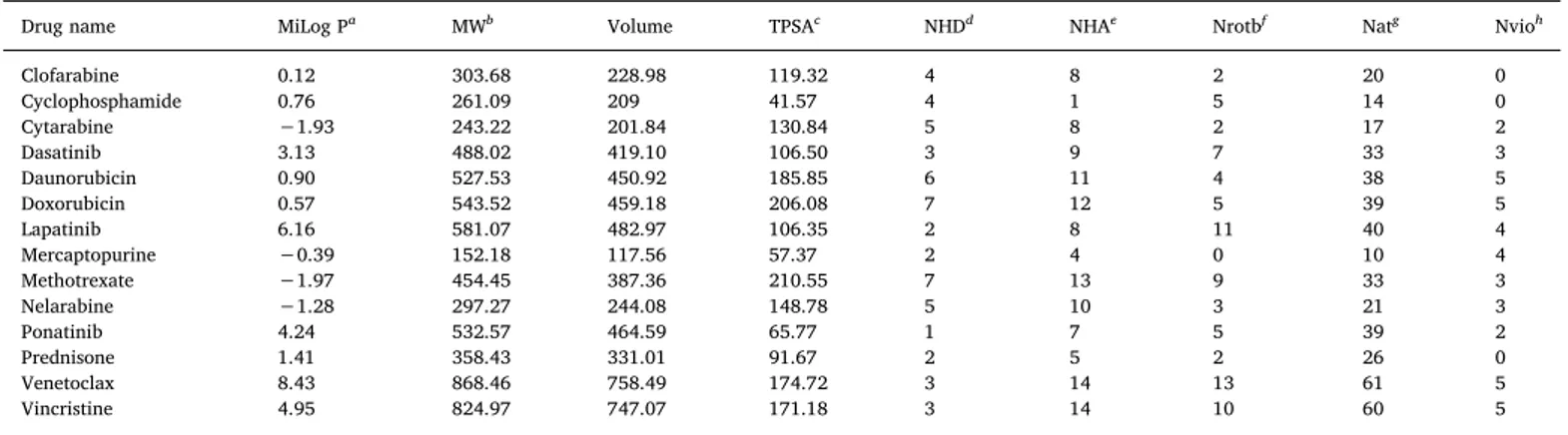
Table 1
Physicochemical properties and drug likeness parameters for the marketed FDA approved drugs used for ALL treatment.aLogarithm of partition coefficient between n-octanol and water (miLog P),bMolecular weight (MW),cTopologicalpolar surface area (TPSA),dNumber of hydrogen bond donors (NHD),eNumber of hydrogen bond acceptors (NHA),fNumber of rotatable bond (Nrotb),gNumber of heavy atoms (Nat),hNumber of violation (Nvio) in terms of drug-likeness criteria.
Drug name MiLog Pa MWb Volume TPSAc NHDd NHAe Nrotbf Natg Nvioh
Clofarabine 0.12 303.68 228.98 119.32 4 8 2 20 0 Cyclophosphamide 0.76 261.09 209 41.57 4 1 5 14 0 Cytarabine −1.93 243.22 201.84 130.84 5 8 2 17 2 Dasatinib 3.13 488.02 419.10 106.50 3 9 7 33 3 Daunorubicin 0.90 527.53 450.92 185.85 6 11 4 38 5 Doxorubicin 0.57 543.52 459.18 206.08 7 12 5 39 5 Lapatinib 6.16 581.07 482.97 106.35 2 8 11 40 4 Mercaptopurine −0.39 152.18 117.56 57.37 2 4 0 10 4 Methotrexate −1.97 454.45 387.36 210.55 7 13 9 33 3 Nelarabine −1.28 297.27 244.08 148.78 5 10 3 21 3 Ponatinib 4.24 532.57 464.59 65.77 1 7 5 39 2 Prednisone 1.41 358.43 331.01 91.67 2 5 2 26 0 Venetoclax 8.43 868.46 758.49 174.72 3 14 13 61 5 Vincristine 4.95 824.97 747.07 171.18 3 14 10 60 5 Table 2
Commonly used “drug-likeness” criteria reported in the literature. MW Molecular weight, NHD Number of hydrogen bond donors, NHA Number of hydrogen bond acceptors, Log P Logarithm of partition coefficient between n-octanol and water, TPSA topological polar surface area, Nat Number of heavy atoms, Nrotb Number of rotatable bond, Nrings Number of rings, Ncarbon Number of carbon atoms.
Criteria C19 Nelarabine
Lipinski 2001 (Lipinski,
2001) MW≤500, NHA≤10mLog P≤4.15, NHD≤5 Yes Yes Ghose 1999 (Ghose et al.,
1999) 160≤MW≤480−0.4≤Wlog P≤5.6 Yes No, 1 violation 40≤MR≤130
20≤Nat≤70 Muegge 2001 (Muegge
et al., 2001) 200≤MW≤600TPSA≤150, Nrotb≤15 Yes No, 1 violation −2≤xLogP≤5
NHD≤5, NHA≤10 Nrings≤7, Ncarbon > 4 Veber 2002 (Veber et al.,
2002) Nrotb≤10, TPSA≤140 Yes No, 1 violation Egan 2000 (Egan et al.,
2000) WLogP≤5.88,TPSA≤131.6 Yes No, 1 violation PAINS 2010 (Baell and
Less number of violation indicates that the design compound has a high chance to be an effective drug, to bind receptors easily, to trans-form through the blood, and to penetrate cell membrane readily. Although obeying/satisfying all druglikeness criteria does not guar-antee to be an effective drug, that means the compound to have a high potency. Our new designed silicon incorporated compound C19 showed to be an appropriate candidate by obeying all of five criteria reported in the literature.
3.3. Evaluation of multi-targeted biological activity
Tumors are resulted from multifaceted process such as modification of genes, change in cell signalling pathways, abnormal molecular
mechanism at multiple levels of tumorigenesis. Therefore, multi-tar-geted approaches have gained growing interest in cancer therapy (Olivo et al., 2010;Li et al., 2014;Hossain et al., 2012;D'Eliseo and Velotti, 2016). Niclosamide, for example, used in the clinical treatment of in-testinal parasite, has been studied as a potential anticancer agent and shown that not only inhibits the related signalling pathways but also targets mitochondria in cancer cells (Li et al., 2014). Curcumin is an-other multi-targeted anti-cancer agents shown to be effective in con-trolling carcinogenesis, inhibition of tumor initiation, inhibition of tumor promotion and progression, induction of tumor cell apoptosis, and retardation of metastasis (Hossain et al., 2012). Based on the pre-ferential multi-targeted approach on cancer therapy, we first evaluated the present approved ALL drugs in terms of their bioactivity scores across the most important drug targets and the results were given in Table 3. Nelarabine showed activity on four drug targets, kinase in-hibitor, GPCR, ion channel modulator, and enzyme inin-hibitor, out of six tested with the highest activity across kinase inhibitor with the score of 0.88 as seen inTable 3. After Nelarabine, its structural analog Clor-afabine follows that with activity on three drug targets. The rest of the two drugs showed a limited bioactivity across the most important drug targets. We, therefore, choosed Nelarabine scaffold as a lead molecule to improve the molecular pharmaceutical properties. Using bioisosteric replacements, we investigated the Nelarabine analogues according to their bioactivity performance (Table 4). According to Molinspiration prediction, the silicon incorporated design compounds C18, C19, C20, and C21 showed the highest activity on the tested target molecules kinase inhibitor, GPCR, ion channel modulator, protease, and enzyme inhibitors. Score of 0.5or higher than that is accepted as a good activity according to the software prediction, while scores between 0.2 and 0.5 are interpreted in moderate activity. Therefore, all four compounds showed less or more protease activity on that Nelarabine showed none. C18 and C20 showed a close value to the limit of good activity for protease inhibitor with the value of 0.48, and 0.47, while C19 and C21 showed a better performance on that with the scores of 0.60 and 0.69.
Table 3
Bioactivity score of the FDA approved anti-ALL drugs across GPCR (G-protein coupled receptor), ion CM (ion channel modulator), kinase, nuclear receptor, protease, and enzyme inhibitor according to Molinspiration software. The va-lues of 0.5 and higher than that are interpreted as good activity and between 0.2 and 0.5 are as moderate activity.
Drug name GPCR Ion CM Kinase Nuclear Protease Enzyme
Clofarabine 1.21 0.39 0.62 −0.79 0.16 1.45 Cyclophosphamide −0.65 −0.38 −0.59 −0.95 −0.33 0.53 Cytarabine 0.73 0.12 0.37 −1.65 0.07 1.12 Dasatinib 0.03 −0.24 0.51 −0.57 −0.27 0.03 Daunorubicin 0.20 −0.15 −0.06 0.29 0.60 0.66 Doxorubicin 0.20 −0.20 −0.07 0.32 0.67 0.66 Lapatinib −0.04 −0.52 0.36 −0.35 −0.21 −0.08 Mercaptopurine −0.87 −0.56 −0.69 −2.71 −1.79 0.04 Methotrexate 0.51 0.23 0.38 −0.38 0.27 0.72 Nelarabine 1.02 0.50 0.88 −1.09 0.00 1.35 Ponatinib 0.36 0.04 0.70 −0.13 0.04 0.13 Prednisone 0.02 −0.12 −0.81 1.00 0.06 0.63 Venetoclax −2.36 −3.42 −3.00 −3.33 −1.92 −2.87 Vincristine −2.00 −3.13 −3.17 −2.94 −1.68 −2.55 Table 4
Bioactivity score of the designed compounds (C1–C23) according to Molinspiration cheminformatics software. GPCR (G-protein coupled receptor), ion CM (ion channel modulator), kinase inhibitor, nuclear receptor, protease, and enzyme inhibitor.
Compd X Y GPCR Ion CM Kinase Nuclear Protease Enzyme
Nel. CH3 NH2 1.02 0.50 0.88 −1.09 0.00 1.35 C1 CH3 CH3 0.94 0.33 0.61 −1.26 0.05 1.11 C2 CH3 H 0.90 0.52 0.73 −1.34 −0.09 1.16 C3 CH3 F 1.10 0.45 0.62 −0.64 0.06 1.26 C4 CH3 Cl 0.95 0.28 0.49 −0.99 −0.14 1.00 C5 CH3 Br 0.83 0.46 0.41 −1.12 −0.16 1.05 C6 CH3 I 1.13 0.45 0.61 −0.89 0.03 1.24 C7 CH3 SH 0.61 0.25 0.43 −1.31 −0.28 0.96 C8 CH3 PH2 0.86 0.45 0.61 −0.91 0.06 1.06 C9 CH3 Si(CH3)3 1.07 0.56 0.68 −0.74 0.46 1.34 C10 NH2 NH2 1.00 0.41 0.82 −1.03 0.12 1.36 C11 H NH2 1.00 0.59 0.72 −1.22 −0.17 1.44 C12 F NH2 0.99 0.48 0.80 −1.03 0.01 1.26 C13 Cl NH2 0.98 0.48 0.80 −1.05 0.00 1.25 C14 Br NH2 0.97 0.46 0.79 −1.06 −0.01 1.25 C15 I NH2 0.98 0.48 0.80 −1.03 −0.00 1.25 C16 SH NH2 0.96 0.48 0.78 −1.03 0.06 1.29 C17 PH2 NH2 1.01 0.39 0.87 −0.96 0.07 1.29 C18 Si(CH3)3 NH2 1.22 0.65 0.85 −0.86 0.48 1.58 C19 Si(CH3)3 Si(CH3)3 1.05 0.54 0.54 −0.62 0.60 1.34 C20 BH2 Si(CH3)3 1.04 0.54 0.61 −0.70 0.47 1.25 C21 B(OH)2 Si(CH3)3 0.96 0.49 0.61 −0.50 0.69 1.37 C22 BH2 NH2 0.98 0.48 0.80 −0.04 0.01 1.26 C23 C2H5 C2H5 0.88 0.30 0.52 −1.07 0.04 0.98
As whole, the four silicon incorporated compounds C18–C21 exhibited better performance than even Nelarabine with five drug targets activity out of six tested. However, remember that C21 violated four out of five druglikeness criteria as discussed in the previoussubsections 3.1and 3.2. So, in addition to the best drug-likeness score discussed before, silicon incorporated compound C19 has also showed a better multi-targeted activity.
3.4. Silicon exposure to human body
Silicon is reported as ‘under investigation’ in clinical trials by DrugBank with the code of DB12982. The only record on the DrugBank website is regarding its Dietary and Nutritional Therapies. Silicon (atomic number 14) has the same number of valency electron with carbon and located just below carbon on the periodic table. After oxygen, silicon takes the second place in the abundancy among other elements in the earth's crust.
We are exposing silicon every day from different sources such as dietary, cosmetics, pharmaceutics, medical implants, and even dust. The natural silicon sources can be count as drinking water, vegetables, fruits, legumes, nuts, even meat and milk (Jugdaohsingh, 2007). For most western countries, daily silicon intake is recorded as over two-fold higher than that of iron and zinc according to the National Academy of Sciences of USA (Copper, 2001). Dietary silicon intake was found to be an important factor for bone heath (Jugdaohsingh, 2007). After our interesting result of silicon bioisostere exhibiting the best bioactivity score, we investigated the literature and found similar and growing tendency of silicon use in medicinal chemistry and in known drug scaffolds. Tacke's and Ramesh's studies worth to mention here as a significant contribution into the field (Bains and Tacke, 2003; Geyer et al., 2015;Ramesh and Reddy, 2017).
3.5. Silicon use in medicinal chemistry
Silicon use in medicinal chemistry has gained growing popularity in last two decades, but the effort is still not enough. Silicon switch with carbon or nitrogen has been widely used in order to increase molecular pharmacological properties due to their structural similarity (Geyer et al., 2015;Ramesh and Reddy, 2017). Some of the differences coming with the silicon incorporation can be ordered as larger bond length, increased lipophilicity, and higher electropositivity as will be discussed in the following subsections.
3.5.1. Larger bond length
When we compared the bond lengths, silicon incorporation makes the bond lengths longer than the lead compound Nelarabine. By re-placing NH2with Si(CH3)3in the position of Y shown inTable 4, bond length of CeN increases from 0, 147 nm to 0.186 nm for CeSi bond. Additionally, replacement of O − (CH3) group with OeSi(CH3)3group in the position of X increases the bond length from 0.143 nm (OeC) to 0.162 nm (OeSi). Silicon's increased bond length upon N+/Si switch was also found beneficial to increase ROR-inhibitory activity by Fujii
and co-workers (Toyama et al., 2015). 3.5.2. Higher lipophilicity
Small lipophilic molecules diffuse trough blood-brain barrier better allowing drug molecules to reach brain and to treat brain related dis-eases. For that purposes, silicon incorporated antibiotics were evaluated in terms of bioactivity and found a significant increase in penetration upon silicon incorporation (Seetharamsingh et al., 2015). Another ex-ample taking advantage of increased lipophilicity upon silicon in-corporation was studied with and approved anticancer natural drug Campothecin. Its several analogs were used in human clinical trials for cancer treatment (Daud et al., 2005;Arnold et al., 2010). Lipophilicity of a molecule which is mostly represented by Log P value is considered as a measure of permeability across cell membrane. Increase in lipo-philicity upon with silicon incorporation was used as an advantage in some drug discovery studies. Docking study of Kagechika and co-workers evaluated estrogenic activity of phenols including silicon in-corporated analogues and found the highest potency in triethylsilicon derivative. Improved estrogenic activity was found to be due to in-creased lipophilicity caused by silicon substitution (Fujii et al., 2012). Lipophilic silicon analogues showed a better performance in cell pe-netration, however, note that could be due to combinational effect of other properties such as changed polarity of the bond, conformational changes. According to Molinspiration software, all of four design compounds listed in Table 5 showed a positive value of lipophilic nature, while Nelarabine showed a negative Log P value of −1.28 in-dicating hydrophilic nature. Among them, C19 showed the highest Log P value of 3.91 indicating much higher lipophilic nature. Leeson's study on the patented compounds with 18 different companies showed that the mean value of the Log P for the patented drugs from the years of 2000 to 2010 ranged from 3.5 to 4.5 (Leeson and St-Gallay, 2011). Based on that, C19 gives a promising result.
3.5.3. Higher electropositivity
Silicon (electronegativity 1.90) is more electropositive element than both carbon and nitrogen with the electronegative value of 2.55 and 3.04 respectively, according to Pauling scale. That causes more polarity on OeSi bond than that of OeC bond of the lead molecule Nelarabine. On the other hand, N+to Si switch brings reversal/decreasing effect on bond polarity. When we compare the total polar surface area of the lead and the design molecules C19 and C20 inTable 5, we see that the de-creasing effect of N+/Si switch has a greater influence on the molecule. While Nelarabine shows TPSA value of 148.78A2, C19 and C20 shows 122.76 A2 upon with decreasing effect of Si on the molecular polar surface area. Higher electropositivity of silicon than nitrogen is used to treat Alzheimer's disease and silicon containing compound was ap-proved for clinical trials (Cutler et al., 1995;Zhu et al., 1995). There-fore, N+/Si or C/Si switch can beused to increase more negative and more positive charge, respectively, to increase the binding energy of the drug with a positive and a negative charge groups on target molecule.
Table 5
Physicochemical properties and drug likeness parameters for our promising design compounds (C18-C21) showed the highest bioactivity and the marketed FDA approved drug Nelarabine.aLogarith of partition coefficient between n-octanol and water (miLog P),bMolecular weight (MW),cTopological polar surface area (TPSA),dNumber of hydrogen bond donors (NHD),eNumber of hydrogen bond acceptors (NHA),fNumber of rotatable bond (Nrotb),gNumber of heavy atoms (Nat), hNumber of violation (Nvio) out of five drug-likeness criteria summarized inTable 2according to SwissADME software. (The SMILES codes of the compounds C18–C21 were provided in the Supplementary Material Table1.)
Drug name MiLog Pa MWb Volume TPSAc NHDd NHAe Nrotbf Natg Nvioh
C18 1.00 355.43 306.75 148.78 5 10 4 24 3/5
C19 3.91 412.60 374.69 122.76 3 9 5 27 0/5
C20 1.00 352.23 330.92 122.76 3 9 4 24 1/5
C21 0.57 384.23 349.53 163.22 5 11 5 26 4/5
3.6. Nelarabine and ara-G
Purine nucleoside analogs such as fluderabine (Keating et al., 1998), cytarabine, pentostatine (Ho et al., 1990) were associated with the clinical evaluation of arabinosyl analog of deoxyguanosine (ara-G). Ara-G is transported into T-cells and intracellularly phosphorilated to ara-Ara-G triphosphate (ara-GTP) which is antineoplastic agent and an inhibitor of DNA synthesis. Similarly, Nelarabine, despite of inactive itself, is used as a pro-drug in chemotherapy for ALL, converts to ara-G by adenosine deaminase as shown inFig. 2which exerts cytotoxic effects. Although this cellular toxicity induced by Nelarabine is used as an advantage, also causes adverse drug reactions. The most important clinical results of Nelarabine is the relation between the accumulation of ara-GTP and the response. For this relation, Nelarabine was sug-gested to be used as prognostic test by evaluating ara-GTP in target tumor population. With this regard, our design compound C19 could still perform the similar function with unstable silyl enol ether bound in the position of X. Therefore, since ara-G could be still produced in vivo, C19 would perform as a prodrug like Nelarabine, in addition to the improved druglikeness properties.
Clinical success of Nelarabine have been reported in indolent leu-kemia (Gandhi et al., 2008), however, dose-dependent toxicities ob-served in phase I and phase II trials must also be taken care. Hemato-logic and nonhematoHemato-logic toxicities such as central and peripheral neurotoxicity have been reported. Somnolence, malaise, fatigue, neu-ropath, and balance abnormalities are the most frequently observed effects (Gandhi et al., 2008;Buie et al., 2007). According to Rockers's review on clinical trials with Nelarabine administration in T-cell ma-lignancy, complete respond changed from 0% to 71% and partial re-spond reported from 8.5% to 35.3% (Roecker et al., 2010). Therefore, improvement on pharmacological properties using Nelarabine scaffold is necessary. Gandhi's phase I study of Nelarabine showed success on patients who did not respond to purine nucleoside analog fluderabine chemotherapy before. For similar reasons, combining therapy of flu-derabine with cytarabine have been tested and validated for ALL, CLL (chronic lymhocytic leukemia), and acute myeloid leukemia (Gandhi et al., 1993;Gandhi et al., 1992). Based on those clinical results, im-provement of existing purine nucleoside analog Nelarabine is ob-ligatory in order to diminish adverse effects, to produce alternative options to patients who might develop Nelarabine refractory/relapse, and to treat very resistance and refractory cancers.
4. Conclusion
In conclusion, we have investigated the existing, FDA approved anti leukemic drugs in terms of their in silico physicochemical properties, five different drug likeness criteria, and biological activities against the most important drug targets GPCR, ion channel modulator, kinase in-hibitor, nuclear receptor ligand, protease, and enzyme inhibitors. Among fourteen marketed drugs, Nelarabine showed the best bioac-tivity score by the highest acbioac-tivity across kinase inhibitor and by tar-geting four out of six drug targets tested. In order to decrease adverse effects and to overcome potential drug resistant and sensitivity pro-blem, we have designed novel therapeutic agents using bioisosteric
replacements into Nelarabine scaffold and evaluating their pharmaco-kinetic properties. From the series of bioisosteric analogues of the marketed anti-leukemia drug Nelarabine (C1–C23), the compound C19 with the incorporation of Si(CH3)3into NH2and CH3groups exhibited even better molecular properties, broader spectrum of bioactivity on five drug targets out of six tested, giving insight to be a better anti-leukemic drug for coming future.
Acknowledgement
This work was supported by the Scientific and Technological Research Council of Turkey, TUBITAK [grant no: 217Z299, 2018]. The author would like to thank Dr. Mustafa Güzel from Istanbul Medipol University for valuable discussion and their suggestion regarding bioi-sosteric groups.
Appendix A. Supplementary data
Supplementary data to this article can be found online athttps:// doi.org/10.1016/j.ejps.2019.04.008.
References
Arnold, S.M., et al., 2010. A phase I study of 7-t-butyldimethylsilyl-10-hydro-xycamptothecin in adult patients with refractory or metastatic solid malignancies. Clin. Cancer Res. 16 (2), 673–680.
Baell, J.B., Holloway, G.A., 2010. New substructure filters for removal of pan assay in-terference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 53 (7), 2719–2740.
Bains, W., Tacke, R., 2003. Silicon chemistry as a novel source of chemical diversity in drug design. Current Opinion in Drug Discovery & Development 6 (4), 526–543.
Buie, L.W., et al., 2007. Nelarabine: a novel purine antimetabolite antineoplastic agent. Clin. Ther. 29 (9), 1887–1899.
Cavelier, F., et al., 2002. Influence of silaproline on peptide conformation and bioactivity. J. Am. Chem. Soc. 124 (12), 2917–2923.
Copper, I., 2001. Dietary Reference Intakes for Vitamin a Vitamin K, Arsenic, Boron, Chromium, Copper, Iodine, Iron, Manganese, Molybdenum, Nickel, Silicon, Vanadium, and Zinc. The National Academies Press, Washington, DC.
Cutler, N.R., et al., 1995. Acetylcholinesterase inhibition by zifrosilone: pharmacokinetics and pharmacodynamics. Clinical Pharmacology & Therapeutics 58 (1), 54–61.
Daud, A., et al., 2005. Phase II trial of karenitecin in patients with malignant melanoma: clinical and translational study. Clin. Cancer Res. 11 (8), 3009–3016.
D'Eliseo, D., Velotti, F., 2016. Omega-3 fatty acids and cancer cell cytotoxicity: implica-tions for multi-targeted cancer therapy. J. Clin. Med. 5 (2), 15.
Egan, W.J., et al., 2000. Prediction of drug absorption using multivariate statistics. J. Med. Chem. 43 (21), 3867–3877.
Fu, R.-G., et al., 2017. Designing multi-targeted agents: an emerging anticancer drug discovery paradigm. Eur. J. Med. Chem. 136, 195–211.
Fujii, S., et al., 2012. Increased hydrophobicity and estrogenic activity of simple phenols with silicon and germanium-containing substituents. J. Med. Chem. 56 (1), 160–166.
Gandhi, V., et al., 1992. Fludarabine infusion potentiates arabinosylcytosine metabolism in lymphocytes of patients with chronic lymphocytic leukemia. Cancer Res. 52 (4), 897–903.
Gandhi, V., et al., 1993. Fludarabine potentiates metabolism of cytarabine in patients with acute myelogenous leukemia during therapy. J. Clin. Oncol. 11 (1), 116–124.
Gandhi, V., et al., 2008. Phase I trial of nelarabine in indolent leukemias. J. Clin. Oncol. 26 (7), 1098–1105.
Geyer, M., et al., 2015. Can silicon make an excellent drug even better? An in vitro and in vivo head-to-head comparison between loperamide and its silicon analogue sila-lo-peramide. ChemMedChem 10 (5), 911–924.
Ghose, A.K., et al., 1999. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1 (1), 55–68.
Gottesman, M.M., 2002. Mechanisms of cancer drug resistance. Annu. Rev. Med. 53 (1),
615–627.
Hanumegowda, U.M., et al., 2010. Phospholipidosis as a function of basicity, lipophili-city, and volume of distribution of compounds. Chem. Res. Toxicol. 23 (4), 749–755.
Ho, A.D., et al., 1990. Pentostatin in refractory chronic lymphocytic leukemia: a phase II trial of the European organization for research and treatment of cancer. JNCI: Journal of the National Cancer Institute 82 (17), 1416–1420.
Holleman, A., et al., 2004. Gene-expression patterns in drug-resistant acute lymphoblastic leukemia cells and response to treatment. N. Engl. J. Med. 351 (6), 533–542.
Holohan, C., et al., 2013. Cancer drug resistance: an evolving paradigm. Nat. Rev. Cancer 13 (10), 714.
Hopkins, A.L., Groom, C.R., 2002. The druggable genome. Nat. Rev. Drug Discov. 1 (9), 727.
Hossain, D., et al., 2012. Curcumin: the multi-targeted therapy for cancer regression. Frontiers in Bioscience (Schol Ed) 4 (1), 335–355.
Jugdaohsingh, R., 2007. Silicon and bone health. J. Nutr. Health Aging 11 (2), 99.
Keating, M., et al., 1998. Long-term follow-up of patients with chronic lymphocytic leukemia (CLL) receiving fludarabine regimens as initial therapy. Blood 92 (4), 1165–1171.
Leeson, P.D., St-Gallay, S.A., 2011. The influence of the ‘organizational factor’ on com-pound quality in drug discovery. Nat. Rev. Drug Discov. 10 (10), 749.
Li, Y., et al., 2014. Multi-targeted therapy of cancer by niclosamide: a new application for an old drug. Cancer Lett. 349 (1), 8–14.
Lipinski, C.A., 2001. Avoiding investment in doomed drugs. Current Drug Discovery 1, 17–19.
Muegge, I., et al., 2001. Simple selection criteria for drug-like chemical matter. J. Med. Chem. 44 (12), 1841–1846.
Olivo, M., et al., 2010. Targeted therapy of cancer using photodynamic therapy in combination with multi-faceted anti-tumor modalities. Pharmaceuticals 3 (5), 1507–1529.
K. L. Prus, et al., Transport and metabolism of 9-β-d-arabinofuranosylguanine in a human T-lymphoblastoid cell line: nitrobenzylthioinosine-sensitive and-insensitive influx, Cancer Res. 50 (6) (1990) 1817–1821.
Ramesh, R., Reddy, D.S., 2017. Quest for novel chemical entities through incorporation of silicon in drug scaffolds. J. Med. Chem. 61 (9), 3779–3798.
Refsgaard, H.H., et al., 2005. In silico prediction of membrane permeability from calcu-lated molecular parameters. J. Med. Chem. 48 (3), 805–811.
Roecker, A.M., et al., 2010. Nelarabine in the treatment of refractory T-cell malignancies. Clinical Medicine Insights: Oncology 4, CMO–S4364.
Seetharamsingh, B., et al., 2015. Design, synthesis, and identification of silicon in-corporated oxazolidinone antibiotics with improved brain exposure. ACS Med. Chem. Lett. 6 (11), 1105–1110.
Toyama, H., et al., 2015. Design and synthesis of novel ROR inverse agonists with a dibenzosilole scaffold as a hydrophobic core structure. Bioorg. Med. Chem. 23 (13), 2982–2988.
Veber, D.F., et al., 2002. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 45 (12), 2615–2623.
Waring, M.J., 2010. Lipophilicity in drug discovery. Expert Opin. Drug Discovery 5 (3), 235–248.
Wishart, D.S., et al., 2017. Drugbank 5.0: a major update to the drugbank database for 2018. Nucleic Acids Res. 46 (D1), D1074–D1082.
Zhu, X.-D., et al., 1995. Effect of MDL 73,745 on acetylcholine and biogenic amine levels in rat cortex. Eur. J. Pharmacol. 276 (1–2), 93–99.