Quantifying interdependent risks in genomic privacy
Tam metin
Şekil
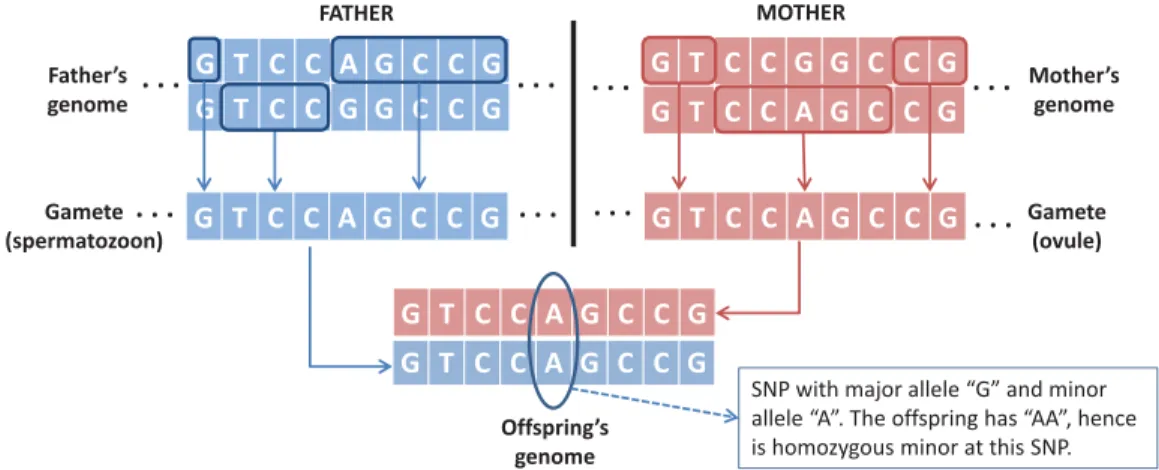
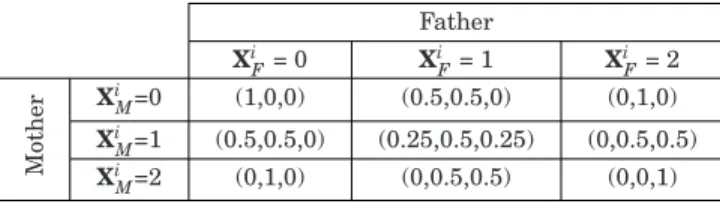

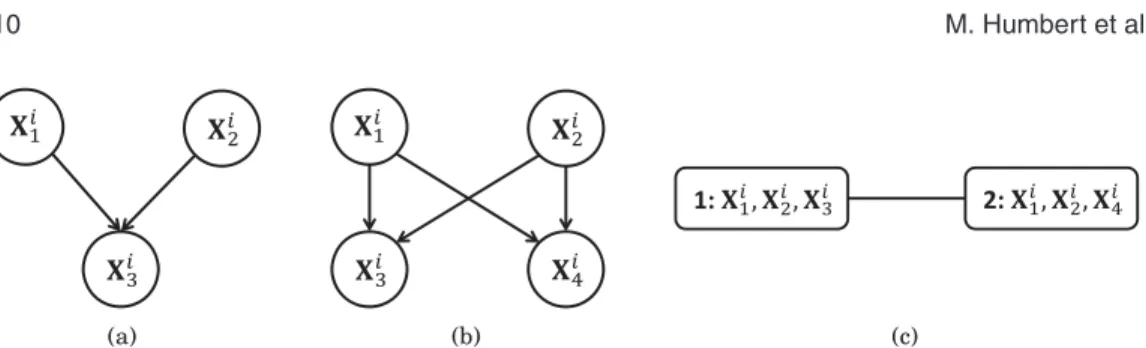
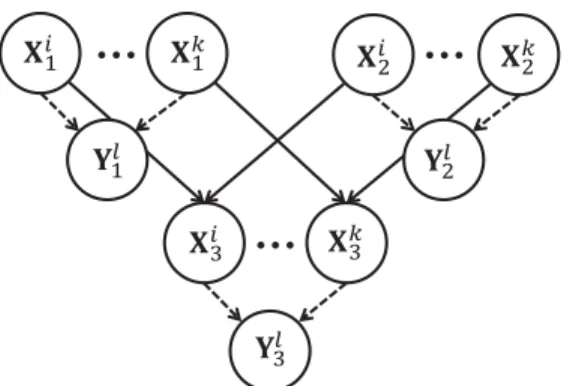

Benzer Belgeler
27 However, our calculations noticeably show that there is no direct correlation between the surface energies of the crystalline slab and the surface area where water reacts or
However, there is a need to investigate whether syllabi of STEM courses offered at EHEA universities are aligned with the STEM education praxis (Kalayci, 2009). Thus, the
The center-right tendency is represented by the Motherland (MP) and the True Path (TPP) parties, and the center-left by the Democratic Left Party (DLP) and the Republican
generations and over time (Finch and Mason, 2007, p. It might occupy a mystical value because of concealing a time which cannot be repeated and perceptions of someone lost.
bir taahhüt, kişilik hakkının aşırı derecede sınırlandırılması anlamına gelmekte olup, İMK md. Bununla birlikte, Federal Mahkeme, söz konusu olaya ilişkin olarak, İBK
A range of very low corruption levels can not be implemented under the presumed guilty rule because the net social marginal benefit of inducing enforcement effort becomes negative
açısından; bu yetkinin kanun olarak mı yoksa karar olarak mı kullanılacağını, beşte üç nitelikli çoğunluk kuralını ve hangi işlemlerin af anlamına geleceğini
The two main features of the interaction to be simulated within our model are (i) the oscillatory energy difference between the ferromagnetic and the antiferromagnetic ground
