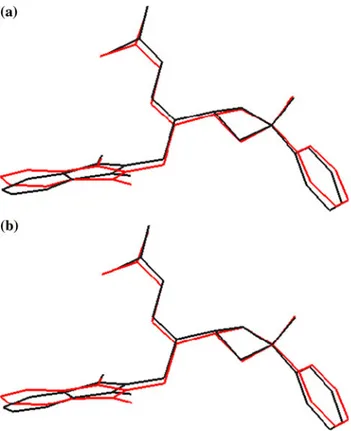
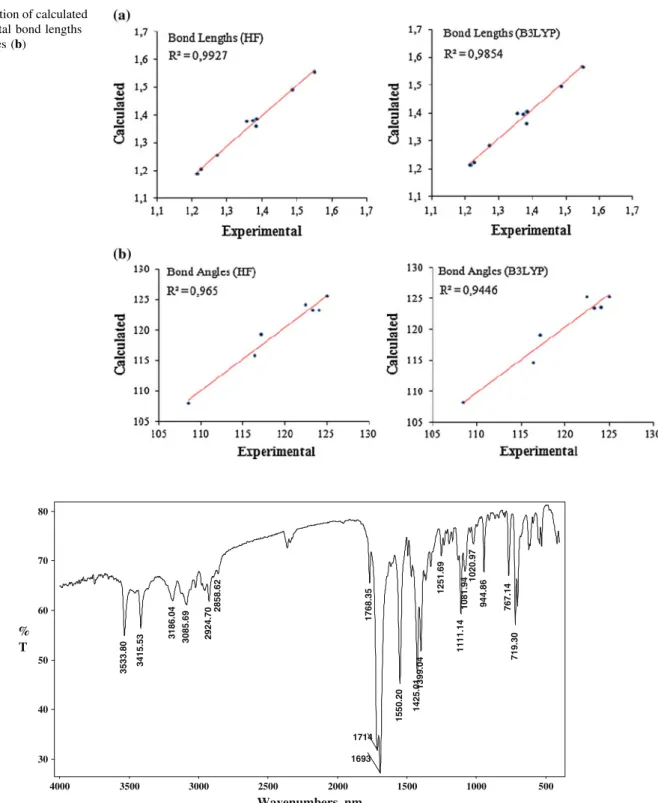
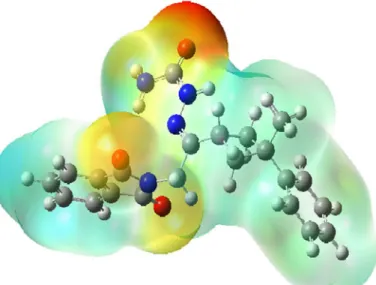
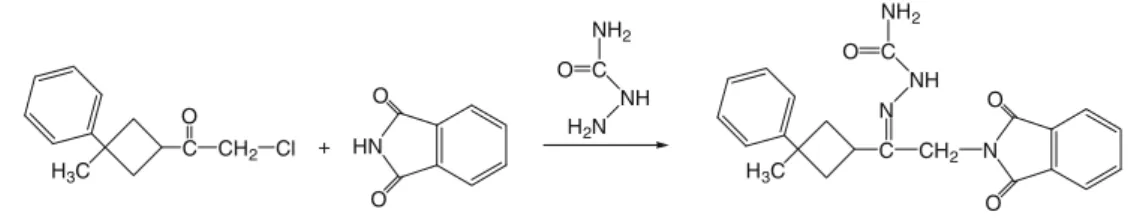
X-ray diffraction and theoretical approach to the molecular structure of (e)-2-(2-(1,3-dioxoisoindolin-2-yl)-1-(3-phenyl-3-methylcyclobutyl)ethylidene) hydrazine carboxamide
Tam metin
Şekil


Benzer Belgeler
Laura Mark’s term “lame infinity” (2010), which ex- plains the methods applied by technology to produce repeating similarities, is clearly reflected in the statements of
Additionally, the predictive role of the skin patch test for corticosteroid hypersensitivity in the clinical response of patients to intranasal corticosteroid therapy should also
A natural language understanding system that takes punctuation into account is the (Constraint Grammar developed by Karlsson and his colleagues [199-1]. This is
Turkish diplomatic passports to Barzani and Talabani, economic aid given to these groups by Turkey, multi-national NGO’s entering the region through Turkey, black market
Eytdi bi-haddur bu tii'atda sevab Geçdi Peygamber yine kıldı cevab Vardı altınçı göge irdi Habtb Ol gök ehlin ne'ydügin gördi Habtb Gördi kim ol gök dahı tolmış
“Orhan Pamuk’u okumaya sondan başladım: Kara Kitap, Beyaz Kale, Sessiz Ev, Cevdet Bey ve Oğulları... Kara Kitap vesilesiyle diğerleri yeniden okunmuş
Kişileri hayatın getirdiği her türlü olumsuzluklara karşı koruması ve bundan daha önem lisi iç h u z u r ve asayişin sağlanarak, cem iyet hayatını ahenkli
