Hetasilin molekülünün atık sulardan uzaklaştırılması
Tam metin
Şekil
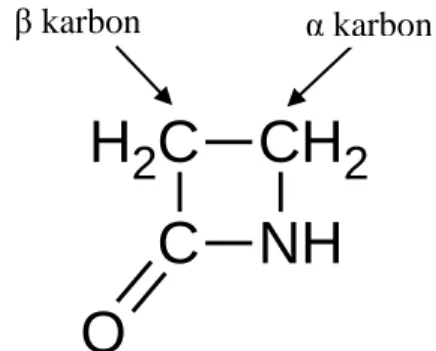
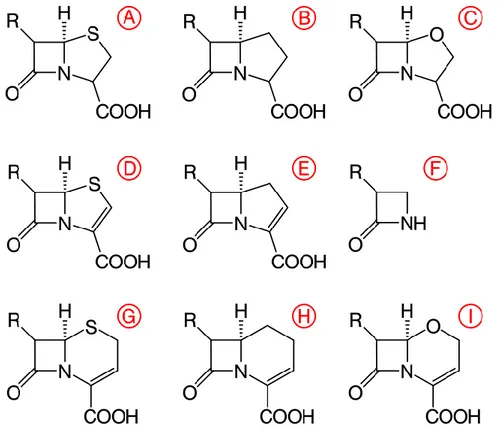
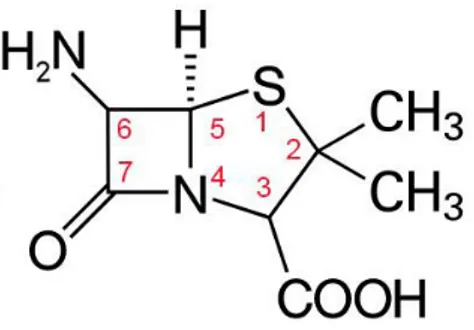
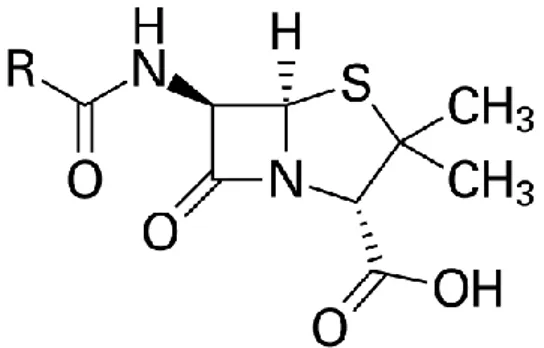
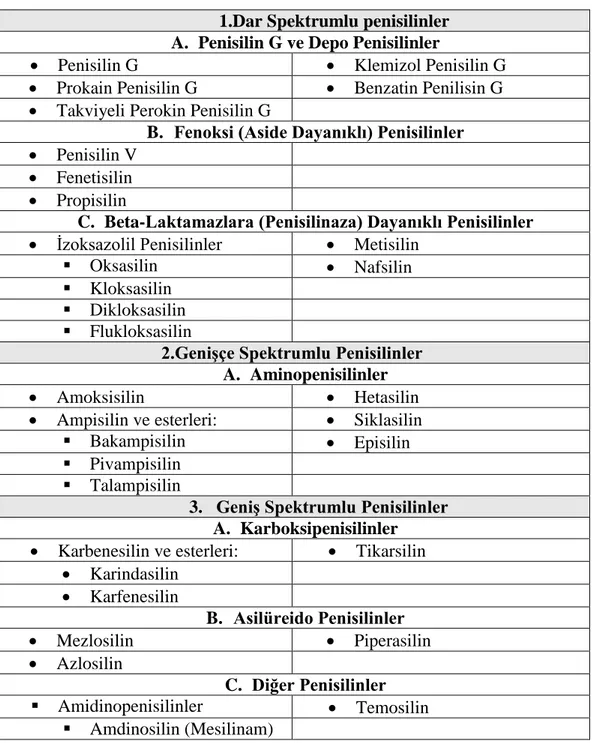
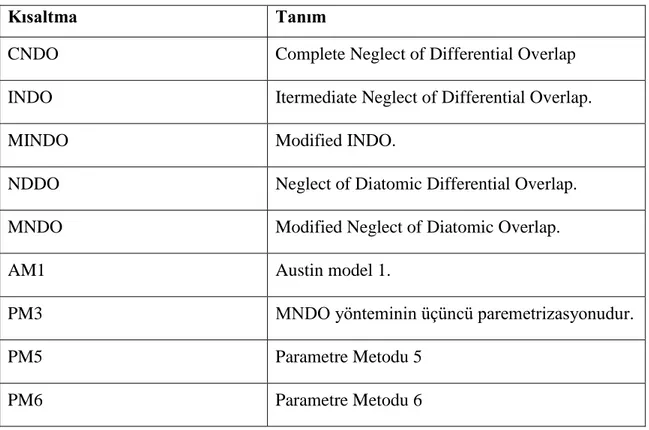
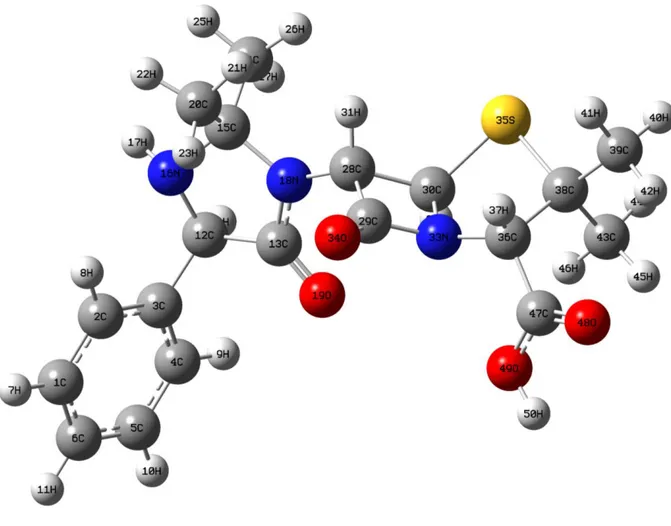
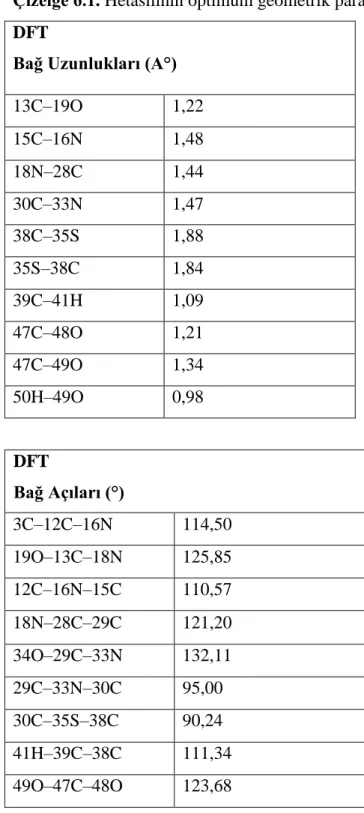

Benzer Belgeler
Dinozorlar dönemi olarak da bilinen ve toplam 180 milyon yıl süren Mezozo- ik Zaman’da (245-65 milyon yıl önce), eldeki fosille- re göre, 500 kadar dinozor türünün
• Histiyosit (Sabit makrofaj): Doku içinde bağ dokusu fibrillerine tutunmuş hareketsiz, yıldız yada iğ biçimli hücrelerdir.. • Serbest makrofajlar: Ara madde içinde
Hafta Bağ tesisi; yer seçimi, anaç ve çeşit seçimi, ekonomik faktörler, arazinin hazırlanması, dikim sistemleri ve fidan dikimi.. Ekonomik faktörler
• Ekonomik koşullar, Arazi hazırlığı • Dikim sistemleri ve dikim sıklığı • Fidan tipinin belirlenmesi.. • Arazinin İşaretlenmesi ve Dikim Çukurlarının
Bunun için, arazinin durumuna bağlı olarak erken sonbahar döneminden başlamak üzere, pulluk tabanının kırılması, derin toprak işleme, toprak örneklerinin alınması ve
PAULSEN Kuvvetli Yüksek Yeterli Yüksek 17(Yüksek) Orta 1613C Kuvvetli Orta Yüksek Zayıf-Orta Düşük Orta 110R Kuvvetli Yüksek Yeterli Çok Yüksek 17(Yüksek) Duyarlı 140
Toprakaltı zararlıları Topraküstü zararlıları Filoksera Nematodlar Salkım güvesi Bağ pirali Tripsler Bağ uyuzu Tripsler Bağ uyuzu Kırmızı örümcekler Maymuncuk
Yeni yüksek fırın için geçen sene Krupp firması ile yapılan mukavele bozulmuş fırının inşası başka bir Alman firmasına ihale edilmiştir. Bu yüzden fırının
