Kraniosinostoz cerrahisi yapılan olguların geriye dönük değerlendirilmesi
Tam metin
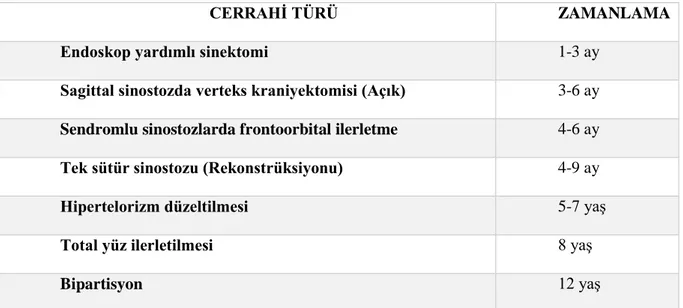
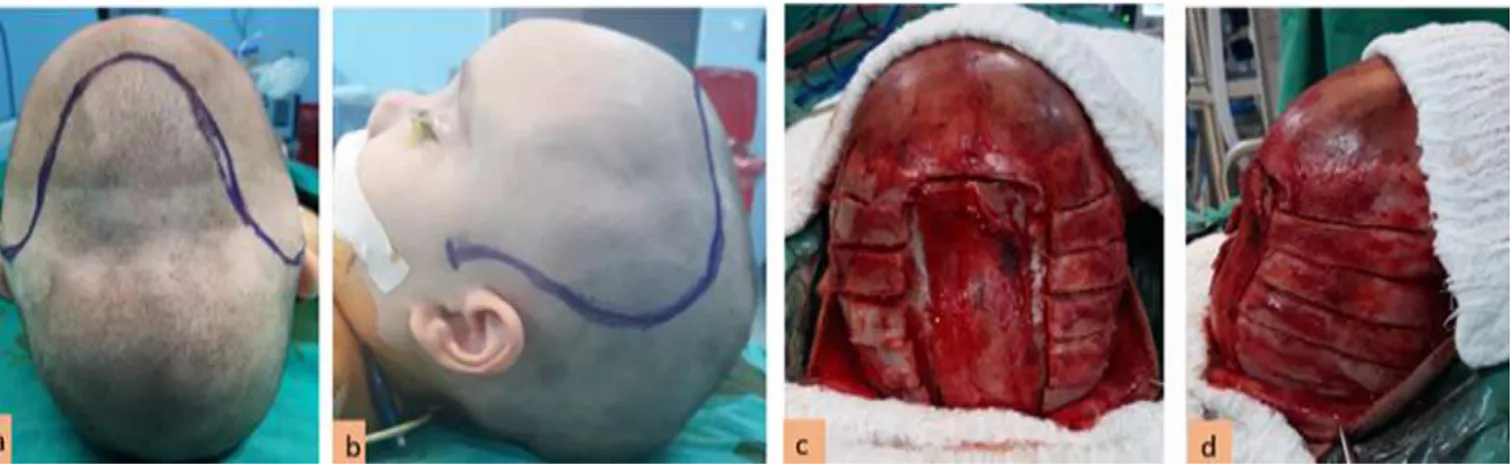
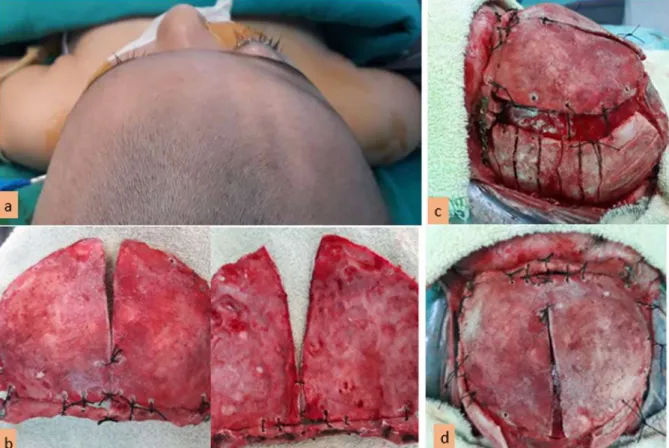
Şekil



Benzer Belgeler
Dört çocuk, on altı torun sahibi Haşan Tepe, hâlâ tarlada tırpan sallıyor; kente gidip yerleşmeyi aklının ucundan bile geçirmemiş. Bahçelerinden kaldırdığı
[r]
Efüzyonlu otitis media, adenotonsiller hipertrofi ve obstrüktif uyku apnesi sık karşılaşılan kulak burun boğaz problemle- ridir.. Mukopolisakkaritlerin östaki tüpü, orta kulak
Objective: To evaluate efficacy and safety of endoscopic sinus surgery (ESS) in pediatric patients with chronic rhinosinusitis (CRS) refractory to
Bu tek ni ğin en bü yük avan taj la rı ola- rak ame li yat sı ra sın da iç ku la ğın ze de len me ris ki nin az olu şu, pro tez mig ras yon oran la rı nın da ha dü şük olu
His to pa to lo jik ke sit ler de in ce kon nek tif stro ma ile ay rıl mış, kis tik di la tas yon gös te ren, kan la do lu ge niş vas kü ler boş luk lar dan mey da na ge len kap
Vakamızda, frontoetmoid mukoselin neden olduğu göz ağrısı ve merkezi ve periferik görme alanında de- fekt bulunmaktaydı.. Diğer tipik göz bulguları
Pamuk Prenses ve Yedi Cüceler (Schneewittchen) masalında yedi cücelerin prensesin ölümünden sonra 3 gün boyunca ağladıklarının bildirilmektedir. Burada yine
