ALG1-CDG: Clinical and Molecular Characterization of 39 Unreported Patients
Tam metin
Şekil
Benzer Belgeler
1 (HO-1) protein expression in the presence or absence of LPS and showed time and dose-dependent inhibition of LPS-induced nitric oxide (NO) production and inducible nitric
The result of our study revealed that the quality of life scales of MS patients with various sleep disturbances were decreased significantly compared to the average of the
Demographical findings and clinical features of the study group CaseGenotypeAge SexSymptom onFamilyCutaneousIDTANDEpilepsySemiologyCT SEN SEGARenalCardiacOphthalmological no
The aim of this study is to estimate the plasma concentrations of fibronectin, AGEs (Advanced Glycosylation End Products), AOPPs (Advanced Oxidation Protein Products) T1DM (Type
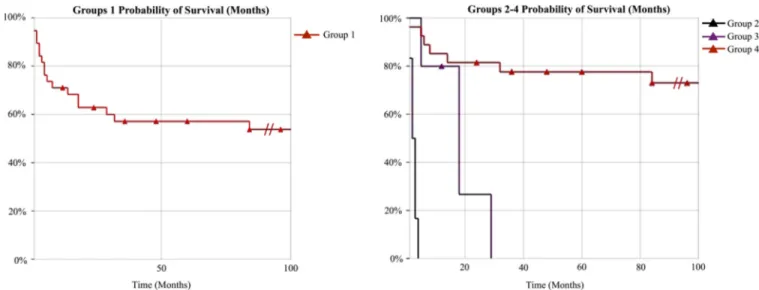
However, in our patient group, since 6 different CDG types were detected in 9 patients and the single type detected in more than 1 patient was PMM2-CDG, it would not be possible
While bone marrow aspiration was performed in 7 patients with pancytopenia, bone marrow culture was studied.. Culture positivity was detected in 4 out of 7
no studies in Turkey on MPS II patients, the objective of this study is to present the genetic and clinical characteristics of patients in Western Turkey with Hunter
Effects of idursulfase enzyme replacement therapy for Mucopolysaccharidosis type II when started in early infancy: comparison in two siblings. Franco JF, Soares DC, Torres LC,
