Koopmans’ springs to life
Ulrike Salzner and Roi Baer
Citation: J. Chem. Phys. 131, 231101 (2009); doi: 10.1063/1.3269030 View online: http://dx.doi.org/10.1063/1.3269030
View Table of Contents: http://jcp.aip.org/resource/1/JCPSA6/v131/i23 Published by the American Institute of Physics.
Additional information on J. Chem. Phys.
Journal Homepage: http://jcp.aip.org/Journal Information: http://jcp.aip.org/about/about_the_journal Top downloads: http://jcp.aip.org/features/most_downloaded Information for Authors: http://jcp.aip.org/authors
Koopmans’ springs to life
Ulrike Salzner1,a兲and Roi Baer2,b兲1
Department of Chemistry, Bilkent University, 06800 Bilkent, Ankara, Turkey 2
Fritz Haber Center for Molecular Dynamics, Institute of Chemistry, The Hebrew University of Jerusalem, Jerusalem 91904, Israel
共Received 27 June 2009; accepted 9 November 2009; published online 16 December 2009兲 The meaning of orbital energies 共OOEs兲 in Kohn–Sham 共KS兲 density functional theory 共DFT兲 is subject to a longstanding controversy. In local, semilocal, and hybrid density functionals共DFs兲 a Koopmans’ approach, where OOEs approximate negative ionization potentials共IPs兲, is unreliable. We discuss a methodology based on the Baer–Neuhauser–Livshits range-separated hybrid DFs for which Koopmans’ approach “springs to life.” The OOEs are remarkably close to the negative IPs with typical deviances of⫾0.3 eV down to IPs of 30 eV, as demonstrated on several molecules. An essential component is the ab initio motivated range-parameter tuning procedure, forcing the highest OOE to be exactly equal to the negative first IP. We develop a theory for the curvature of the energy as a function of fractional occupation numbers to explain some of the results. © 2009 American Institute of Physics.关doi:10.1063/1.3269030兴
An attractive feature of the Hartree–Fock共HF兲 theory is the interpretation inspired by Koopmans’1 that negative oc-cupied orbital energies 共OOEs兲 approximate ionization po-tentials共IPs兲 of atoms and molecules. Whether this is true in density functional theory共DFT兲 is subject to a longstanding controversy.2–13It has been established that the negative en-ergy of the highest occupied molecular orbital 共HOMO兲 in the Kohn–Sham 共KS兲 DFT is equal to the first IP.6,14 This holds also for generalized Kohn–Sham 共GKS兲 approaches.15,16As for the meaning of the deeper KS OOEs, opinions vary: From “there is no physical meaning at all”3to “exact KS negative OOEs are close to IPs even for low-lying energy levels.”9
Numerical and theoretical evidences demonstrate that exact KS OOEs are excellent approximations to quasiparticle energies obtained by Green’s function methods.17–20 This sharply contrasts the failure of local/semilocal and hybrid DFs: HOMO energies underestimate first IPs by several elec-tron volts.21,22As demonstrated below, this holds for deeper OOEs as well.4,5 One problem of approximate DFs is the presence of spurious self-interaction14,23 关mostly exchange not canceling Hartree self-repulsion 共SR兲2兴 artificially in-creasing OOEs of localized orbitals.12 Mitigating self-interaction2,24–31 can be achieved with range-separated hybrids共RSHs兲, applied within a GKS formalism.21,32–41 In RSHs, the exchange energy splits into two: an explicit long-range orbital 共erf共␥r兲/r兲 and a local/semilocal short-range 共erfc共␥r兲/r兲 components.␥ is the range-parameter共in a0−1兲. In this letter, we discuss the use RSHs for estimating of IPs. The specific RSH we use combines the Baer– Neuhauser–Livshits共BNL兲 RSH DF21and the ab initio mo-tivated range-parameter tuning procedure共␥-tuning, where␥ is the range-parameter兲. Our tuning procedure enforces the exact GKS condition −HOMO= IP⌬SCF共Refs.16and21兲
as-sociating quantities from differing charge states of the sys-tem. Such procedure is implemented as a line search and was discussed in Ref.21, requiring few additional self-consistent field 共SCF兲 ground state calculations of the neutral and its cation. We present the IP predictions and compare to stan-dard DFs. Range-parameter tuning procedures ameliorate several prominent failures of common DFT applications: dis-sociation of radicals,42,43 localization of charge in weakly interacting systems,44charge-transfer excitations,45and band gaps in solids.46 Finally, we develop a theory that partially explains the success of tuned RSHs and the failures of local, semilocal, and hybrid DFs.
We first compare IPs calculated using traditional DFs and tuned-␥ BNL 共BNLⴱ兲 for small molecules. We check two ways for estimating IPs: 共1兲 “Koopmans’:” set IPk= −Ne−k+1.共2兲 “⌬SCF/TD:” the first IP 共IP1兲 is estimated using a⌬SCF procedure 共the cation neutral SCF energy dif-ference兲 and IPk+1= IP1+ hk共k=1,2,...兲, where hkare the time-dependent GKS equations’ cation excitation energies. We test how calculated IPs compare to experimental IPs and how the two methods compare to each other. To avoid basis-set truncation errors we used Dunning’s correlation-consistent polarized valence-quadruple-zeta 共CC-PVQZ兲 basis-sets47 throughout. Geometries of N2, O2, F2, water 共H2O兲, ammonia 共NH3兲, formaldehyde 共CH2O兲, and formic acid 共HCOOH兲 were optimized with the coupled-cluster singles doubles共CCSD兲 method, with the HF method, with KS-DFT at the local spin-density approximation 共LSDA兲 level, with the gradient corrected BP86共Becke exchange and Perdew’s 86 correlation DF兲, with Becke’s hybrid, B3LYP, with B3P86 and the B3P86%–30% hybrid and with BNL. In the latter, ␥ was adjusted for each neutral system to repro-duce −HOMO= IP⌬SCF. Coupled cluster IPs were calculated as single points with disconnected triples关CCSD共T兲兴 on the CCSD geometries. BNL calculations used QCHEM 3.2.48 Other DF calculations used GAUSSIAN 03.49 We used the random-phase approximation option for the time-dependent
a兲Electronic mail: [email protected]. b兲Electronic mail: [email protected].
DFT 共TDDFT兲 calculations, except for F2 and O2, where only Tamm–Dancoff option converged.
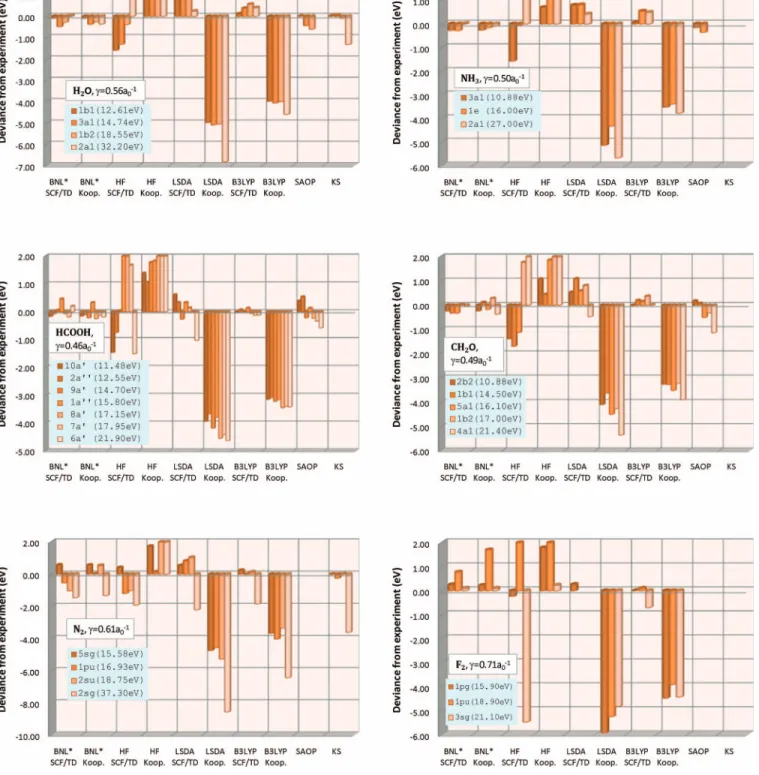
In Fig.1we compare several estimates with experimen-tal vertical IPs for H2O, NH3, HCOOH, CH2O, N2, and F2. More detailed data are given in the supporting information. The F2results deserve special attention, as the cation ground 共excited兲 state is doubly degenerate where the hole can be in g+or ing−共u+or inu−兲 orbitals. Thus, there are two types of hole transitions:g +→ u + org +→ u −
which have the same energy but TDDFT breaks this degeneracy due to functional deficiency. The TD data for IP2 of F2 in Fig.1 refers to the
g+→u+transition. We summarize the results in Fig.1: 共1兲 HF theory deviances vary in the range ⫺2 and 2 eV for
all systems. Koopmans’ deviances are positive while ⌬SCF/TD IPs are usually negative. For N2+, HF spu-riously predicts ⌸u ground state symmetry instead of ⌺g. For F2, HF predicts⌺g symmetry for the first ex-cited cation state instead of⌸u. KS and GKS methods avoid such large qualitative errors.
共2兲 LSDA: Koopmans’ deviances are large 共⫺5 to ⫺8 eV兲 and non-uniform because SR is larger in localized
FIG. 1. Deviance of negative OOEs and SCF/TD energies relative to vertical IPs derived from experiment data共Refs.50–52兲 for several molecules. KS and
SAOP OOEs data are taken from Ref.9.
orbitals.12⌬SCF/TD deviances are about 1 eV. 共3兲 B3LYP: Koopmans’ deviances are still large, ⫺3 to
⫺4 eV, but more uniform than in LSDA due to smaller SR.⌬SCF/TD IPs deviances are small 共0.5 eV兲. 共4兲 BNLⴱ: IPs, whether computed by Koopmans’ or
⌬SCF/TD methods, exhibit low deviances. For the larger molecules the deviances are 0.3 eV or less while for diatomics some OOEs have larger deviances共note: the N2 experimental IP4 is insecure and the peak is multiconfigurational53兲. BNLⴱdeviances are small even for deep valence orbitals 共IPs of ⫺20 to ⫺33 eV兲. BNLⴱ OOEs are also close to true KS OOEs and slightly superior to statistical averaging of orbital potentials 共SAOP兲 results.9 The performance for -orbitals is better than for -orbitals. BNLⴱ Koopmans’ and⌬SCF/TD IPs are close for both outer and inner orbitals. For core orbitals of water and N2 BNL OOEs are 5 and 8 eV higher than true KS orbitals, ⬃25 eV above experimental IPs.9
共5兲 In LSDA, B3LYP, and BNL the SCF/TD predictions for the IPs are all reasonably good in accordance with previously established results 共provided the states do not have double excitation character兲.
One can improve the LSDA and B3LYP Koopmans’ IP predictions by adding a constant shift IP1共⌬SCF兲+H. This works better for B3LYP than for LSDA共because of SR兲: the IP deviances of the first three orbital energies in H2O are fairly constant in LSDA/B3LYP,⬃−4 eV to −5 eV, but that of the compressed 2a1 orbital, deviates by 2 eV in LSDA and by 0.6 eV in B3LYP. In BNLⴱ this effect is unnoticeable.
We now provide a theory to help explain some of the numerical results. Following Refs. 22 and54 we highlight the concept of the curvature of the energy Egs 共in KS/ GKS/HF theories兲 with respect to fi, the occupation number of the ith molecular orbitali共r兲. The importance of curva-ture stems from Janak’s theorem55i=Egs/fi, so:
Egs关N − 1;i兴 − Egs关N兴 =
冕
1 0i共fi兲dfi, 共1兲
where Egs关N兴 is the ground state energy of the N=2NH closed shell electron system共NHis the index of the HOMO兲 and Egs关N−1;i兴 is the hole-constrained DFT ground state of the N − 1 electron system with a hole at the ith orbital. This “fully relaxed” excited state energy for the cation approxi-mates the variational excited state DFT method;56so the left hand side of Eq.共1兲approximates IPNH+1−i. When the curva-ture共2Egs/fi
2兲=共
i/fi兲 is zero, as it is for the HOMO in exact KS or GKS theories, then the right-hand side of Eq.共1兲 equals −i and this is approximately equal to the relevant IPNH+1−i. When the curvature is positive, Eq. 共1兲 yields −i共1兲⬍IPNH+1−i, as found in calculations with approximate DFs for the HOMO energy, discussed in Refs.22and54.
We now give an expression for the full curvature matrix: Cmi⬅共2Egs/fmfi兲=共m/fi兲=共i/fm兲. For clarity, we assume closed shell molecules and we suppress the spin des-ignation for the orbitalsi共r兲 and OOEs. The Hamiltonian Hˆ is given by␦Egs/关␦i共r兲兴=Hˆi共r兲 and the density matrix is
兺ifii共r兲i共r
⬘
兲. The KS/GKS equations assert that Hˆi =ii and since fi are parameters in Hˆ , we have57k共r兲/fi=兺j⫽kkj−1A共jk兲 i j共r兲, where A共jk兲i ⬅具j兩Hˆ /fi兩k典 and kj=k−j; in particular Cmi= A共mm兲 i . Now, Hˆ /fi not only creates the matrix elements A共jk兲i but also depends on them and from this,
A共nm兲i =
兺
jk共R−1兲
共nm兲共jk兲W共jk兲共ii兲, 共2兲 where the W matrix corresponds to linear response kernel, W共jk兲共mn兲=
冕冕
d3rd3r⬘
冋
冉
1r − r
⬘
+ fXC共r,r⬘
兲冊
j共r兲n共r⬘
兲 − u共兩r − r⬘
兩兲j共r⬘
兲n共r兲册
m共r⬘
兲k共r兲. 共3兲 This matrix arises from the dependency of the molecular orbitals on the occupation numbers. R−1is the inverse of the total response matrix,R共jk兲共mn兲=␦共jk兲共mn兲+nm
−1共f
n− fm兲W共jk兲共mn兲. 共4兲 In Eq. 共3兲, fXC共r,r
⬘
兲=␦vXC关n兴共r兲/␦n共r⬘
兲 is the XC kernel andvXC关n兴共r兲 is the KS or GKS XC potential. In HF theoryfXCis zero. The function u共r兲, describing orbital exchange, is zero in KS theory, 1/r in the HF theory, and its choice char-acterizes the kind of GKS theory used: u共r兲=/r 共where 0⬍⬍1兲 for hybrid DFs 共in B3LYP =0.2兲 and u共r兲=erf共␥r兲/r for RSH 共BNL兲 DFs 共in this latter case vXC关n兴共r兲 is dependent on the range-parameter␥ as well兲.
The relation in Eq.共2兲 is exact but difficult to analyze. To simplify, we neglect the off-diagonal elements of the ma-trix R in Eq.共4兲, neglecting all W共mn兲共jk兲in Eq.共4兲except for same-pair interactions, when 共mn兲=共kj兲. In this case A共nm兲i = W共nm兲共ii兲/R共mn兲共mn兲and in particular,
Cmi= A共mm兲
i ⬇ W
共mm兲共ii兲. 共5兲
Applying this result to orbitaliitself we find the curvature element Cii⬇W共ii兲共ii兲, i.e.,
Cii⬇
冕冕
d3rd3r⬘
关u¯共兩r − r⬘
兩兲 + fXC共r,r⬘
兲兴i共r兲2i共r⬘
兲2, 共6兲 where u¯共r兲=r−1− u共r兲. For the HF theory both fXCand u¯共r兲 vanish and thus Cii⬇0, a result corroborated for i=NH in calculations, showing small curvature, only slightly negative.31,54,58,59 For local/semilocal hybrid DFs, u¯共r兲=共1 −兲/r 共=0, local/semilocal and =0.2, B3LYP兲 and fXC共r,r
⬘
兲⬀␦共r−r⬘
兲, leading to W共ii兲共ii兲dominated by positive Hartree SR energy for orbital i共r兲. This gives significant positive curvature, within semilocal DFs as corroborated by numerical calculations.31,54,58–60We are not aware of calcu-lations for hybrids. Positive curvature grows for localized orbitals as these have large SR. Thus LSDA IPs have larger deviances than B3LYP, which has partial cancellation of SR. In the exact KS theory, the nonlocal fXC共r,r⬘
兲 kernel cancels SR and the curvature should be small. In GKS-RSH theories,which are intermediate between HF and local KS theories, self-interaction is small and the rule that Cii⬇0 holds well as seen in numerical calculations.31,54 In BNL curvatures are small but not exactly zero, thus requiring ␥-tuning to have the initial slope ␦Egs/␦fH兩fH=1 equal to the average slope −IP1= Egs关N兴−Egs关N−1兴.
Summarizing, we gave numerical and theoretical evi-dence suggesting that ab initio motivated ␥-tuned BNL en-ables that of Koopmans’ approach using OOEs to approxi-mate IPs to good accuracy. The tuning procedure was found essential for quantitative predictions in other “tough” prob-lems for DFT and TDDFT.16,43–46
Supplemental material is available:61Table with IPs for N2, O2, F2, H2O, NH3, CH2O, and HCOOH at various the-oretical levels.
This work was supported by the European Union 7, framework project Unam-Regpot 共Contract 203953兲 and TŰBITAK 共Contract 109T426兲, the Bilkent University, and by the Israel Science Foundation共Contract 962/06兲.
1T. C. Koopmans,Physica共Amsterdam兲 1, 104共1934兲. 2J. P. Perdew and A. Zunger,Phys. Rev. B 23, 5048共1981兲. 3J. P. Perdew and M. R. Norman,Phys. Rev. B 26, 5445共1982兲. 4J. P. Perdew and M. Levy,Phys. Rev. Lett. 51, 1884共1983兲.
5A. R. Williams and U. von Barth, in Theory of the Inhomogeneous
Elec-tron Gas, edited by S. Lundqvist and N. H. March共Plenum, London,
1983兲.
6C. O. Almbladh and U. Von Barth,Phys. Rev. B 31, 3231共1985兲. 7P. G. Parr and W. Yang, Density-Functional Theory of Atoms and
Mol-ecules共Oxford University Press, New York, 1989兲.
8R. Stowasser and R. Hoffmann,J. Am. Chem. Soc. 121, 3414共1999兲. 9D. P. Chong, O. V. Gritsenko, and E. J. Baerends,J. Chem. Phys. 116,
1760共2002兲.
10O. V. Gritsenko and E. J. Baerends,J. Chem. Phys. 117, 9154共2002兲. 11O. V. Gritsenko, B. Braïda, and E. J. Baerends,J. Chem. Phys. 119, 1937
共2003兲.
12T. Körzdörfer, S. Kümmel, N. Marom, and L. Kronik,Phys. Rev. B 79, 201205共2009兲.
13S. Kümmel and L. Kronik,Rev. Mod. Phys. 80, 3共2008兲.
14J. P. Perdew, R. G. Parr, M. Levy, and J. L. Balduz,Phys. Rev. Lett. 49, 1691共1982兲.
15A. Seidl, A. Gorling, P. Vogl, J. A. Majewski, and M. Levy,Phys. Rev. B 53, 3764共1996兲.
16R. Baer, E. Livshits, and U. Salzner, Annu. Rev. Phys. Chem. 61, 85 共2010兲.
17L. S. Cedérbaum, G. Hohlneicher, and W. Niessen,Chem. Phys. Lett.18, 503共1973兲.
18L. S. Cederbaum, G. Hohlneicher, and S. Peyerimhoff,Chem. Phys. Lett. 11, 421共1971兲.
19J. D. Doll and Wp. Reinhard,J. Chem. Phys. 57, 1169共1972兲. 20L. J. Sham and W. Kohn,Phys. Rev. 145, 561共1966兲.
21E. Livshits and R. Baer,Phys. Chem. Chem. Phys. 9, 2932共2007兲. 22A. M. Teale, F. De Proft, and D. J. Tozer,J. Chem. Phys. 129, 044110
共2008兲.
23D. J. Tozer and N. C. Handy,J. Chem. Phys. 109, 10180共1998兲. 24A. D. Becke,J. Chem. Phys. 98, 5648共1993兲.
25M. E. Casida, K. C. Casida, and D. R. Salahub,Int. J. Quantum Chem.
70, 933共1998兲.
26M. E. Casida and D. R. Salahub,J. Chem. Phys. 113, 8918共2000兲. 27S. Ivanov and R. J. Bartlett,J. Chem. Phys. 114, 1952共2001兲. 28L. Veseth,J. Chem. Phys. 114, 8789共2001兲.
29D. J. Tozer,J. Chem. Phys. 119, 12697共2003兲.
30F. Della Sala and A. Gorling,J. Chem. Phys. 118, 10439共2003兲. 31A. J. Cohen, P. Mori-Sanchez, and W. T. Yang,J. Chem. Phys. 126,
191109共2007兲.
32A. Savin, in Recent Advances in Density Functional Methods Part I, edited by D. P. Chong共World Scientific, Singapore, 1995兲, p. 129. 33H. Iikura, T. Tsuneda, T. Yanai, and K. Hirao,J. Chem. Phys.115, 3540
共2001兲.
34T. Yanai, D. P. Tew, and N. C. Handy,Chem. Phys. Lett.393, 51共2004兲. 35J. Toulouse, A. Savin, and H. J. Flad,Int. J. Quantum Chem. 100, 1047
共2004兲.
36R. Baer and D. Neuhauser,Phys. Rev. Lett. 94, 043002共2005兲. 37I. C. Gerber and J. G. Angyan,Chem. Phys. Lett. 415, 100共2005兲. 38M. J. G. Peach, T. Helgaker, P. Salek, T. W. Keal, O. B. Lutnaes, D. J.
Tozer, and N. C. Handy,Phys. Chem. Chem. Phys. 8, 558共2006兲. 39O. A. Vydrov and G. E. Scuseria,J. Chem. Phys. 125, 234109共2006兲. 40Y. Zhao and D. G. Truhlar,J. Phys. Chem. A 110, 13126共2006兲. 41J. D. Chai and M. Head-Gordon,J. Chem. Phys. 128, 084106共2008兲. 42R. Baer, E. Livshits, and D. Neuhauser,Chem. Phys. 329, 266共2006兲. 43E. Livshits and R. Baer,J. Phys. Chem. A 112, 12789共2008兲. 44E. Livshits, R. Baer, and R. Kosloff,J. Phys. Chem. A 113, 7521共2009兲. 45T. Stein, L. Kronik, and R. Baer,J. Am. Chem. Soc. 131, 2818共2009兲. 46H. R. Eisenberg and R. Baer,Phys. Chem. Chem. Phys. 11, 4674共2009兲. 47J. T. H. Dunning,J. Chem. Phys. 90, 1007共1989兲.
48Y. Shao, L. F. Molnar, Y. Jung, J. Kussmann, C. Ochsenfeld, S. T. Brown, A. T. B. Gilbert, L. V. Slipchenko, S. V. Levchenko, D. P. O’Neill, R. A. DiStasio, Jr., R. C. Lochan, T. Wang, G. J. O. Beran, N. A. Besley, J. M. Herbert, C. Y. Lin, T. Van Voorhis, S. H. Chien, A. Sodt, R. P. Steele, V. A. Rassolov, P. E. Maslen, P. P. Korambath, R. D. Adamson, B. Austin, J. Baker, E. F. C. Byrd, H. Dachsel, R. J. Doerksen, A. Dreuw, B. D. Dunietz, A. D. Dutoi, T. R. Furlani, S. R. Gwaltney, A. Heyden, S. Hirata, C.-P. Hsu, G. Kedziora, R. Z. Khalliulin, P. Klunzinger, A. M. Lee, M. S. Lee, W. Liang, I. Lotan, N. Nair, B. Peters, E. I. Proynov, P. A. Pieniazek, Y. M. Rhee, J. Ritchie, E. Rosta, C. D. Sherrill, A. C. Simmonett, J. E. Subotnik, H. Lee Woodcock III, W. Zhang, A. T. Bell, and A. K. Chakraborty,Phys. Chem. Chem. Phys. 8, 3172共2006兲. 49M. J. Frisch, G. W. Trucks, H. B. Schlegel et al.,
GAUSSIAN 03, Gaussian, Inc., Wallingford CT, 2003.
50A. W. Potts and W. C. Price,Proc. R. Soc. London, Ser. A 326, 181 共1972兲.
51K. L. Nixon, W. D. Lawrance, and M. J. Brunger,Chem. Phys. Lett.474, 23共2009兲.
52H. Van Lonkhuyzen and C. A. De Lange,Chem. Phys. 89, 313共1984兲. 53J. A. Nichols, D. L. Yeager, and P. Jorgensen,J. Chem. Phys. 80, 293
共1984兲.
54A. J. Cohen, P. Mori-Sanchez, and W. T. Yang,Phys. Rev. B77, 115123 共2008兲.
55J. Janak,Phys. Rev. B 18, 7165共1978兲.
56M. Levy and A. Nagy,Phys. Rev. Lett. 83, 4361共1999兲. 57S. T. Epstein,Am. J. Phys. 22, 613共1954兲.
58A. Ruzsinszky, J. P. Perdew, G. I. Csonka, O. A. Vydrov, and G. E. Scuseria,J. Chem. Phys. 126, 104102共2007兲.
59J. P. Perdew, A. Ruzsinszky, G. I. Csonka, O. A. Vydrov, G. E. Scuseria, V. N. Staroverov, and J. Tao,Phys. Rev. A 76, 040501共2007兲. 60A. J. Cohen, P. Mori-Sanchez, and W. T. Yang,Science 321, 792共2008兲. 61See EPAPS supplementary material at http://dx.doi.org/10.1063/
1.3269030 for experimental and computed vertical ionization energies 共eV兲 for various molecules.