i T.C.
EGE ÜNİVERSİTESİ TIP FAKÜLTESİ
İÇ HASTALIKLARI ANABİLİM DALI
KRONİK MİYELOPROLİFERATİF HASTALIK TANILI HASTALARDA CALR TİP 1 VE CALR TİP 2 GEN MUTASYONU DURUMUNUN
SAPTANMASI VE KLİNİK İLE İLİŞKİSİ
UZMANLIK TEZİ Dr. MİRAY YAMAN
DANIŞMAN:
Prof. Dr. GÜRAY SAYDAM
İZMİR 2016
ii Önsöz
İç hastalıkları uzmanlık eğitimim sürecinde bilgi, deneyimleri ve hoşgörüsü ile her zaman yanımda olan başta Anabilim Dalı Başkanımız Prof. Dr. Fehmi Akçiçek’e, Uzmanlık tezim ve eğitim hayatımın her aşamasında bana destek olan ve yardımlarını eksik etmeyen tez hocam Prof. Dr. Güray Saydam’a,
Uzmanlık eğitimim boyunca bilgi ve tecrübelerini esirgemeden paylaşan başta Prof. Dr. Fahrettin Oksel’e, Prof. Dr. F. Rüçhan Uslu’ya, Prof. Dr. A. Ömer Özütemiz’e ve tüm diğer kıymetli hocalarıma,
ardiyolo i, nfeksiyon astalıkları ve Göğüs astalıkları Anabilim Dalı’nda rotasyoner olarak geçirdiğim süre zarfında bilgilerinden faydalandığım başta Prof. Dr. Abdullah Sayıner ve kıymetli hocalarıma,
yıl boyunca uyum içinde çalıştığımız, birlikte çalışmaktan zevk aldığım asistan arkadaşlarıma, değerli uzman hekimlere, klinik ve poliklinik hemşire ve çalışanlarına ayrı ayrı en içten teşekkürlerimi sunarım.
Tez çalışmam sırasında Tıbbi Biyolo i Anabilim Dalı çalışanlarına gösterdikleri bilimsel destek ve ilgileri için en içten teşekkürlerimi sunarım.
Bu süreçte tüm fedakarlıkları ile hep yanımda olan ve bana güç veren aileme çok teşekkür ederim.
Dr. Miray YAMAN İzmir- 2016
iii Proje Bilgisi
ge Üniversitesi İç astalıkları ematolo i Polikliniğine başvuran daha önce 11.04.2007 tarih ve 07-3/3 sayılı protokol numarası ile protrombin ve faktör V Leiden gen mutasyonu bakılan kronik miyeloproliferatif hastalık ( MP ) tanılı hastaların 76’sının örnekleri -70°C’de saklanıp ge Üniversitesi Tıp Fakültesi Tıbbi Biyolo i Anabilim Dalında çalışılmıştır. Bu çalışma ge Üniversitesi Pro e Araştırma ve Destekleme Fonu (Proje No: 2015-TIP-052) tarafından desteklenmiştir.
iv İÇİNDEKİLER Önsöz ...ii Proje Bilgisi.……….…..………...….iii İçindekiler ………..………...……...iv Kısaltmalar ...v
Tablo Dizini ...vii
Şekil Dizini ………...………..…...viii
Özet ...ix
Abstract ( İngilizce özet) ...xi
1. Giriş ve Amaç ...…………..………....…………...………..………..………..…….…1
2.Genel Bilgiler ...4
2.1 Kronik Miyeloproliferatif Hastalıklar...4
2.1.1 Polisitemia Vera...7
2.1.2 Esansiyel Trombositoz...10
2.1.3 Primer Miyelofibroz ………..……...13
2.2 Kronik Miyeloproliferatif Hastalıklarda Tanımlanmış Mutasyonlar ...16
3.Materyal ve Metod ...25
4.Bulgular...33
5.Tartışma …...35
v KISALTMALAR
AML: Akut miyeloid lösemi ASXL: Additional sex-comb like Ca++: Kalsiyum
CALR: Calreticulin
CBL: Casitas B-lineage lymphomaproto-oncogene CNL: ronik nötrofilik lösemi
DIPSS-pluss: İnternasyonel Prognostik Skorlama Sistemi DNA: Deoksiribonükleik asit
DNMT3A: DNA methyltransferase 3A EPO: Eritropoietin
EPO-R: ritropoietin reseptörü ER: Endoplazmik retikulum ET: Esansiyel Trombositemi EZH: Enhancer of Zeste homolog G-CSF: Granülosit stimüle edici faktör
GM-CSF: Granülosit-makrofa stimüle edici faktör Hb: Hemoglobin
HMGA2: High mobility groupAT-hook 2 IDH: İzositrat dehidrogenaz
IKZF: Ikaros Familiy Zinc Finger JAK: Janus Kinaz
JH: JAK homoloji domain KML: ronik miyelositer lösemi KMML: ronik miyelomonositer lösemi KMPH: ronik miyeloproliferatif hastalık LAP: Lökosit alkalen fosfataz
LDH: Laktat dehidrogenaz
LNK: Lenfosit spesific adaptor protein MCV: ırmızı hücre hacmi
MDS: Miyelodisplastik sendrom
MPL: Miyeloproliferatif lösemi virüs onko en V (thrombopoietin receptor) MPN: Miyeloproliferatif Neoplazi
vi
PIAS: Protein Inhibitors of Activated STATs PMF: Primer miyelofibroz
PRC: Policomb represiv complex PV: Polisitemia vera
PCR: Polimeraz zincir reaksiyon RA: Refrakter anemi
RS: Ring sideroblast
RUNX: Runt related transcription factor SH2: SRC homoloji 2 domaini
SOCS: Suppressor of cytokine signaling
STAT: Signal Transducers and Activators of Transcription TET: Ten eleven translocation
TGF: Transforming growth factor TP53: Tumor protein p53
TPO: Trombopoietin TYK 2: Tirozin kinaz 2 WHO: Dünya Sağlık Örgütü vWF: Von Willebrand Faktör
vii TABLO DİZİNİ
Tablo 1. Miyeloid neoplazmların 2008 (W O) sınıflandırma şemas ………..….5
Tablo 2. Hematolojik malignitelerde edinilen gen mutasyonlarının frekans ve dağılımı………...…6
Tablo 3. 2001-2015 yılları arasında W O MPN tanı kriterlerinin evrimi ...8
Tablo 4. Polisitemia vera olgularında risk sınıflaması ...……….……..….…...9
Tablo 5. PV tedavi yönetimi …………...………...….….10
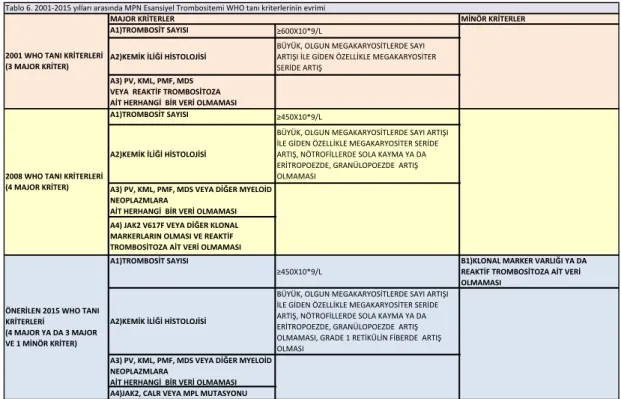
Tablo 6. 2001-2015 yılları arasında MPN sansiyel Trombositemi W O tanı kriterlerinin evrimi ………..………...11
Tablo 7. 2001-2015 yılları arasında MPN Primer Miyelofibrozis W O tanı kriterlerinin evrimi ………..……….…....15
Tablo 8. Calreticulin gen mutasyon analizlerinde kullanılan cihazlar…………...….26
Tablo 9. Calreticulin gen mutasyonlarının analizlerinde kullanılan kimyasal maddeler………..………..26
Tablo 10. Sanger sekans reaksiyonu bileşenleri ………...…………..…...……..32
Tablo 11. Sanger sekans reaksiyonu için PCR döngüleri ………...……33
Tablo 12. astaların cinsiyet dağılımı ………...………..…………....3
Tablo 13. astaların yaş gruplarına göre dağılımı ……….…...34
Tablo 14. linik parametrelerinin tanılara göre dağılımı ………...………….…....35
Tablo 15. İ retikülin lif artışı dercelerinin tanılara göre dağılımı ……..…….………..36
Tablo 16. epatomegali ve splenomegali varlığının tanılara göre dağılımı ….…...…...36
Tablo 17. Tromboembolik olay sıklığının tanılara göre dağılımı …………..……….…37
Tablo 18. Tanı gruplarına göre ortalama izlem süreleri ………..……….…...37
Tablo 19. JA 2 mutasyon sıklığının gruplara göre dağılımı ………..……….…...38
viii ŞEKİL DİZİNİ
Şekil 1.JA 2 proteini domain yapısı………..….……...….16
Şekil 2. JAK-STAT Yolu ………...…………..…………..…….….18
Şekil 3. CALR tip 1 ve tip 2 mutasyonu ………..…………..…..23
ix ÖZET
Kronik Miyeloproliferatif Hastalık Tanılı Hastalarda CALR Tip 1 ve CALR Tip 2 Gen Mutasyonunun Saptanması ve Klinik İle İlişkisi
Amaç: ronik miyeloproliferatif hastalıklar, kemik iliğinde bir ya da daha fazla
miyeloeritroid hücre serisinde kontrolsüz proliferasyon ve periferik kanda artmış olgun hücre sayısı ile karakterize edilen hastalıklar grubudur. Philadelphia (-) klasik kronik miyeloproliferatif hastalıklar ( MP ) başlıca polistemia vera (PV), esansiyel trombositemi ( T) ve primer miyelofibroz (PMF) olarak gruplandırılır. 2005 yılında tanımlanan JA 2V617F mutasyonu ile PV hastalarının %95-98’inde, T ve PMF hastalarının %50’sinde klonal gelişimi göstermek mümkün olmuştur. Daha sonra PV’de JAK2 exon-12 mutasyonları, PMF ve T’de MPL mutasyonları tanımlanmıştır. KMPH hastalarında tanımlanan diğer mutasyonlardan biri de CALR mutasyonudur. ndoplazmik retikulum üzerinde ve içinde Ca++
(kalsiyum) bağlayan şaperon olarak bulunan proteini kodlayan bu mutasyonlar ile hematopoetik hücrelerdeki STAT sinyal yolağı aktive edilerek hücre çoğalması uyarılır. İlk kez 2013 yılında tanımlanan CALR mutasyonları birçok hematolo ik malignite, lenfoid hastalıklar ve solid tümörlerde de gösterilmiştir. CALR mutasyonunun sıklığı T’de %50-70, PMF’de %60-90 oranında bulunmuştur. CALR mutasyonunun hastalığın prognozu ve sağkalım üzerine etkilerine yönelik veriler mevcut olup bu konudaki çalışmalar tüm dünyada devam etmektedir. Bu çalışmada ge Üniversitesi Tıp Fakültesi astanesi ematolo i Polikliniği’nde takip edilmekte olan Ph(-) KMPH hastalarında CALR mutasyonunun belirlenmesi ve CALR mutasyon varlığının hastaların klinik bulguları üzerine etkileri araştırılmıştır.
Gereç ve Yöntem: Çalışmaya 2008 W O kriterlerine göre 35 ET, 1 PMF, 4 POST-PV MF, 4 POST-ET MF ve 32 POST-PV olmak üzere toplam 76 Philadelphia (-) KMPH hastası dahil edildi. astaların demografik özellikleri, klinik ve laboratuar bilgileri kayıt altına alınmıştır. 11.04.2007 tarih ve 07-3/3 sayılı protokol numarası ile protrombin ve faktör V Leiden gen mutasyonu bakılan KMPH tanılı hastaların 76’sının -70°C’de saklanan venöz kandan izole edilmiş DNA’larını kullanarak dizi analizi yöntemiyle CALR mutasyonu çalışılmıştır. İstatistiksel analizlerin değerlendirilmesinde Pearson ki-kare testi ,sıklıklar, ve sağkalım (survival) analizleri kullanılmıştır.
Bulgular: Olguların ortalama yaşı 60,8±13,7’dir. Yaş grupları olarak
değerlendirildiğinde hastaların %6 ,5’i 0-65 yaş aralığındadır. em yaş hem de cinsiyet açısından gruplar arasında fark bulunmamaktadır.PMF tanılı olgularda retikülin lif artışı daha yüksek bulunmuştur ve istatiksel olarak anlamlı saptandı. (p=0,001)
x astalık evresi ile korele olan splenomegali PMF ve PV olgularında daha yüksek saptandı. (p=0,001) Olguların %19,7’sinde başta MI,SVO, Budd,Chiari ve DVT olmak üzere tromboembolik olay saptandı, ancak gruplar arası istatiksel fark bulunmadı. 1 PV olgusunda AML’ye transformasyon görüldü. Sağkalımları açısından gruplar arası anlamlı fark saptanmadı.JA 2 mutasyonu değerlendirilen 5 olguda T’de %20, PV’de %3 , ve PMF’de %33,3 JA 2V617F mutasyonu olduğu görüldü.(p>0,05) CALR tip 1 ve CALR tip 2 mutasyonu açısından değerlendirilen 76 olguda mutasyon saptanmadı.
Sonuç: JAK2 ve MPL mutasyonu (-) olan olgularda en sık saptanan mutasyon olarak
bildirilen ( yaklaşık %80) CALR 52-bp delesyon p.L367fs*46 (Tip 1 CALR mutasyonu) ve 5-bp insersiyonu p.K385fs*47 (Tip 2 CALR mutasyonu) olgularımızda saptanmamıştır. Literatürde bu iki mutasyondan tip 1 CALR mutasyonu %45-50, tip 2 CALR mutasyonu ise %32- 1 görülmektedir. Ancak 50’den farklı mutasyon bu alanda tanımlanmıştır. astalığın patogenezinin aydınlatılması, klinik ve sağkalım ile ilişkilendirilmesini sağladığı gibi ve bu konuda yapılacak çalışmalar ile hedefe yönelik tedaviler açısından yeni çalışmaların da önünü açacaktır.
xi ABSTRACT
CALR Type 1 and CALR Type 2 Gene Mutations Determination and Clinical Relationship with Chronic Myeloproliferative Disorders
Objective: Chronic myeloproliferative diseases are a group of diseases characterized
by uncontrolled proliferation of one or more lines of myeloerythroid cells in bone marrow and increased number of mature or immature cells in the peripheral blood. Philadelphia (-) classic chronic myeloproliferative diseases (CMPD) mainly polycythemia vera (PV), essential thrombocythemia (ET) and primary myelofibrosis (PMF) are grouped. JAK2V617F mutation identified in 2005 have been able to demonstrate clonal growth in 95-98% of PV and 50% of ET and PMF patients. Then JAK2 exon 12 mutations in PV and MPL mutations have been identified in ET and PMF. Other mutations identified in patients with CMPD is also CALR mutation. Cell proliferation is stimulated by activating STAT signaling pathway in hematopoietic cells with these mutations encoding a protein which Ca++ binding chaperone in the endoplasmic reticulum. For the first time identified in 2013 CALR mutations are shown in several hematological malignancy, lymphoid diseases and solid tumors. The frequency of CALR mutations are 50-70% in ET and 60-90% in PMF. The data is present on the effects of CALR mutations on prognosis of disease and survey and studies continues on this subject all over the world. The aim of this study is to investigate the prevalence of CALR mutations at Ph (-) CMPD patients following in outpatient clinic of haematology at Ege University Faculty of Medicine Hospital and their effects on clinical symptoms of patients with mutations in the presence of CALR.
Material and Method: 35 ET, 1 PMF , 32 PV , 4 Post-PV MF and 4 Post-ET MF that
a total of 76 Philadelphia (-) CMPD patients were included to the study according to 2008 WHO criteria. The demographic characteristics of the patients, clinical and laboratory data were recorded. CALR mutations were studied at 11.04.2007 date and 07-3/3 of the protocol number of the viewed with prothrombin and factor V Leiden gene mutation diagnosis of 76 CMPD patients of stored at -70°C venous blood were isolated DNA by sequence analysis method using. Pearson chi-square test, frequencies and survival analysis was used for the evaluation of istatistical analysis.
Results: The mean age of the patients was 60,8 ± 13,7. Considering the age group 40-65 years of age is 64.5% of patients. There is no difference between the groups of both age and also in terms of gender. Reticulin fibers increase in the diagnosis of FMF patients was higher and was statistically significant.(p=0,001) Splenomegaly, which is correlated with stage of the disease has been detected higher in the PMF and PV cases.
xii (p=0,001) In 19.7% of cases, thromboembolic events, including particularly myocardial infarction, cerebrovascular disease, Budd-Chiari and deep vein thrombosis was detected, but statistical differences were not found between groups. AML transformation was seen in the one of PV patients. There was no significant difference between the groups in terms of survival. JAK2V617F mutation was seen in JAK2 mutation has been evaluated in 45 patients with 20% ET, 34.4% PV and 33.3% PMF.(p>0,05) The mutation was not found in 76 cases evaluated in terms of CALR type 1 and CALR type 2 mutations.
Conclusions: JAK2 and MPL mutations (-) patients with the most frequent mutations reported (approximately 80%) CALR 52-bp deletion p.L367fs*46 (Type 1 CALR mutation) and 5-bp insertion p.K385fs*47 (Type 2 CALR mutation) were not detected in our patients. CALR type 1 and type 2 mmutations show the rate of respectively 45-50% and 32-41% in the literature. However, different from the 50 mutations have been described. Studies on this subject describe of the pathogenesis as to be associated with clinical and survival will pave the way for new studies of targeted therapies.
1 1. GİRİŞ VE AMAÇ
ronik miyeloproliferatif hastalıklar ( MP ) hematopoetik hücrelerin bir veya daha fazlasında hücresel çoğalma ile birlikte genellikle farklılaşma ve olgunlaşmanın devam ettiği klonal hematopoetik bozukluklardır. Sonuçta genellikle olgunlaşmış hücrelerde artışa bağlı hücre sayısında artış ile karakterizedir. MP genetik olarak transforme olmuş hematopoetik hücrelerden köken alır ve diferansiasyon, efektif miyelopoez kapasitesi vardır.1
KMPH terimi ilk kez 1951 senesinde William Dameshek tarafından öne sürülmüş olup, birbirine klinik ve biyolo ik benzerlikleri olan dört klasik miyeloproliferatif hastalığı (polisitemia vera (PV), esansiyel trombositoz ( T), primer miyelofibroz (PMF) ve kronik miyeloid lösemi ( ML) ) tarif etmek icin kullanılmıştır.2
Daha ender görülen kronik nötrofilik lösemi, kronik eosinofilik lösemi, sistemik mastositoz, atipik ML gibi hastalıklar daha sonradan MP kapsamına alınmıştır.3
KML yaklaşık 30 yıldan uzun süredir bilinen ve ‘Philadelphia kromozomu’ ve bunun onkogeni BCR-ABL1 pozitif klondan gelişen bir neoplazidir. Ön planda lökositoz ile kendini gösterir, akut blastik faza geçiş %15 civarındadır.1
BCR-ABL1 negatif MP grubunda en sık görülenler PV, T ve PMF olup 2005 yılında JA -2 mutasyonu tanımlanması ile patogenezde rol oynayan genetik faktörler üzerine çalışmalar artmıştır. 2005 yılında tanımlanan JA 2V617F mutasyonu ile PV hastalarının %95-98’inde, T ve PMF hastalarının %50’sinde klonal gelişimi göstermek mümkün olmuştur.4-8
Daha sonra PV’de JA exon-12 mutasyonları, PMF ve T’de MPL mutasyonları tanımlanmıştır.9
Ancak T ve PMF’de hastaların yaklaşık %35- 0’ında JA 2 ve MPL mutasyonları negatif bulunmuştur.
lampfl ve arkadaşları tarafından 2013’de JA 2 negatif T ve PMF hastalarda calreticulin (CALR) somatik gen mutasyonu tanımlanmıştır. CALR geni 9 ekzondan oluşur ve 19p13.2 de lokalizedir.10
CALR şaperon aktivitesi ve kalsiyum homeostazında rol alan, hücre çoğalması, farklılaşması ve apopitozis gibi immüno enik hücre ölümü gibi birçok fonksiyonu olan yüksek derecede korunmuş bir endoplazmik retikulum ( R) proteinidir. R içinde ve dışında bulunarak lipid ve protein sentezi, Ca+2 depolanması, posttranslasyonel modifikasyon gibi hücre zarı, sitoplazma, nükleus ve ekstrasellüler matrikste birçok hücresel fonksiyona sahiptir.11-12
2013’den günümüze ekzon 9’da 50’den farklı CALR mutasyonu tanımlanmıştır ve hepsi de frameshift mutasyon niteliğindedir.13 n sık saptanan ( yaklaşık %80) mutasyon CALR 52-bp delesyon p.L367fs*46 (Tip 1 CALR mutasyonu) ve 5-bp insersiyonu p.K385fs*47 (Tip 2 CALR mutasyonu) olarak saptanmıştır.10 JAK2 ve MPL negatif ET
2 hastalarının %67 ve PMF hastalarının %88’inde bu mutasyonlar gösterilmiştir.10
Nangalia ve arkadaşlarının yaptığı çalışmada ise 151 MP , 13 5 hematolo ik malignite, 1517 diğer kanserler ve 550 kontrol grubunda CALR mutasyonu araştırılmış, JAK2 negatif KMPH hasta grubunda %70-8 oranında saptanmış. Ayrıca MDS-RARS (miyelodisplastik sendrom-refrakter anemi ring sideroblastlı), CMML (kronik miyelomonositer lösemi), KML-RS ( ML ring sideroblastlı), AML (akut miyeloid lösemi), sistemik mastositoz, eozinofilik hastalıklar, lenfoid kanserler, solid tümörler ve kontrol grubunda da 1 farklı CALR mutasyonu saptanmıştır. 52 bp delesyon(CALR Tip 1) mutasyonunun 5 bp insersiyonuna (CALR Tip 2) göre sıklığı daha fazla bulunmuştur.13
JA 2, MPL ve CALR mutasyonları <%10 hastada negatif (triple negatif) bulunmuştur. Yine bazı hastalarda JA 2 ve CALR mutasyonlarının beraber olduğu gösterilmiştir.14-16
CALR (+) T hastalarında JA 2 (+) olanlara göre hemoglobin ( b) ve lökosit sayılarında azalma, trombosit sayısında artış ve kümülatif tromboz insidansında azalma olduğu görülmüştür. astalık progresyon insidansında JA 2 (+) T hastalarına göre azalmaolmasına rağmen lampf ve arkadaşlarına göre sağkalımda uzama saptanmış, ancak Nangalia ve arkadaşlarına göre JA 2 (+) lere göre bir fark saptanmamıştır.10,13,17
yapılan bir çalışmada T hastalarında CALR mutasyonu daha erken yaş, erkek cinsiyet, trombosit sayısının daha yüksek seyretmesi ve hemoglobin, lökosit sayılarında ve tromboz riskinde azalma ile ilişkili gösterilmiştir.18
CALR (+) PMF hastalarında JA 2 ve MPL pozitif hastalara göre daha erken yaş, trombosit sayısının daha yüksek seyretmesi, düşük DIPSS-plus (Dinamik İnternasyonel Prognostik Skorlama Sistemi) skoru, anemi ve lökositoz insidansında azalma, transfüzyon bağımlılığı olma olasılığı daha az olduğu saptanmıştır.15
JAK2 ve CALR Tip 2 mutasyonu (+) olan PMF hastalarına göre Tip 1 CALR mutasyonu pozitifliği sağkalımda anlamlı artış ve iyi prognoz olarak değerlendirilmiştir.19
Sıklığı, hastalığın seyri ve prognoza etkisi nedeniyle CALR ekzon 9 mutasyonlarının MP tanı algoritmasında olması gerektiği ilk defa 201 yılında önerildi.20
2015 yılı Ağustos ayından itibaren W O (Dünya Sağlık Örgütü) PV, ET ve PMF tanı kriterlerini güncelleyerek CALR mutasyonlarına yer verdi. Tüm dünyada bu konuda gittikçe artan sayıda çalışma mevcut olup ülkemizde bu yönde yapılmış yeterli çalışma yoktur.
3 Bu çalışmanın amacı ge Üniversitesi Tıp Fakültesi astanesi ematolo i Polikliniği’nde takip edilmekte olan ML dışı MP hastalarında CALR mutasyonunun belirlenmesidir. CALR mutasyonu dizi analizi yöntemiyle tespit edilmiştir. astaların demografik özellikleri, klinik ve laboratuar bilgileri kayıt altına alınmıştır. Venöz kandan DNA izolasyonu yapılarak sanger sekans dizi analizi yöntemiyle CALR mutasyonu çalışılmıştır. CALR mutasyon varlığının hastaların klinik bulguları üzerine etkileri araştırılmıştır.
4
2. GENEL BİLGİLER
2.1 Kronik Miyeloproliferatif Hastalıklar
ronik Miyeloproliferatif astalıklar (KMPH) pluripotent hematopoetik kök hücredeki bozukluk nedeni ile ortaya çıkan, hematopoetik hücre dizi öncü hücrelerinin anormal proliferasyonu sonucu granülosit, eritrosit veya trombosit sayısında artış ile birlikte sekonder miyelofibrozis ve nadiren de lösemik dönüşümle karakterize klonal hematolo ik hastalıklardır.21
Polisitemia vera (PV), Vaquez tarafından 1892’de, primer miyelofibrozis (PMF) ile yakın zamanlarda, esansiyel trombositoz ( T) ise 1930’larda tanımlandı ( ,5,6). 1951’de William Dameshek, bu hastalıkların klinik bulguları arasında ortak noktaları fark ederek kronik miyeloid lösemi ( ML) ve diğer nadir bozukluklarla beraber, birbiriyle ilişkili bir hastalık spektrumu oluşturduğunu ileri sürdü ve “miyeloproliferatif bozukluklar” terimini ortaya attı (7).
2008 yılında W O tarafından MP adlandırması Miyeloproliferatif Neoplaziler (MPN) olarak değiştirildi. MPN geleneksel olarak ‘klasik’ ve ‘atipik’ olarak sınıflandırılır.. Klasik MPN ; Philadelphia (Ph) translokasyonu ve BCR-ABL füzyon geni taşıyan kronik miyeloid lösemi ( ML) ve Ph-negatif polisitemia vera (PV), esansiyel trombositemi ( T) ve primer miyelofibrozisten (PMF) oluşur. Atipik MPN grubunda ise daha nadir görülen kronik miyelomonositik lösemi, uvenil miyelomonositik lösemi, kronik nötrofilik lösemi, kronik bazofilik lösemi, kronik eozinofilik lösemi, hipereozinofilik sendrom, sistemik mastositoz ve sınıflandırılmamış MPN yer alır (Tablo 1).22,23
1960 yılında Ph translokasyonu ve BCR-ABL füzyonu tanımlanması ile ML patogenezinde gerçekleşen aydınlanma sonrasında ancak 2005 yılından sonra Ph(-) hastalarda JA 2V617F mutasyonu tanımlanmasıyla PV ve diğer klasik MPN’lerin patogenezinin açıklanmasına yönelik çalışmaların önü açılmıştır. JA 2V617F mutasyonu ile PV hastalarının %95-98’inde, T ve PMF hastalarının %50’sinde klonal gelişimi göstermek mümkün olmuştur.4-8
Daha sonra PV’de JA exon-12 mutasyonları, PMF ve T’de MPL mutasyonları tanımlanmıştır.9
2013 yılında T ve PMF hastalarında JA 2V617F mutasyonundan sonra 2. en sık görülen mutasyon olan CALR mutasyonu tanımlanmıştır ve prognostik belirteç olarak kullanlmaya başlanmıştır.10,25
5
Tablo 1.Miyeloid neoplazmların 2008 (WHO) sınıflandırma şeması22
CALR geni 9 ekzondan oluşur ve 19p13.2 de lokalizedir.10
CALR şaperon aktivitesi ve kalsiyum homeostazında rol alan, hücre çoğalması, farklılaşması ve apopitozis gibi immüno enik hücre ölümü gibi birçok fonksiyonu olan yüksek derecede korunmuş bir endoplazmik retikulum ( R) proteinidir. R içinde ve dışında bulunarak lipid ve protein sentezi, Ca+2 depolanması, posttranslasyonel modifikasyon gibi hücre zarı, sitoplazma, nükleus ve ekstrasellüler matrikste birçok hücresel fonksiyona sahiptir.11-12
6 2013’den günümüze ekzon 9’da 0’dan farklı CALR mutasyonu tanımlanmıştır ve hepsi de frameshift mutasyon (çerçeve kayması) niteliğindedir.13
n sık saptanan (yaklaşık %80) mutasyon CALR 52-bp delesyon p.L367fs*46 (Tip 1 CALR mutasyonu) ve 5-bp insersiyonu p.K385fs*47 (Tip 2 CALR mutasyonu) olarak saptanmıştır.10 Bu mutasyonlarla karboksi-terminal ucunda (C-Terminal) yeni bir aminoasit sekansının meydana gelmesine neden olur. CALR’in c-MPL ve JA 2 sinyal yolaklarını aktive ettiği düşünülmektedir. Mutant CALR ekspresyonu ile Ba/F3 hücrelerinde (murine cellline) IL-3 (İnterlökin-3) bağımlılığı olmadan JA 2 sinyal yolu aktiflenir. Böylece sitokin bağımsız hücre çoğalması ve büyümesi indüklenir.10,13,45,46
Bu da KMPH patogenezindeki etkilerinden biri olarak tanımlanmıştır.
Günümüzde T T2 (T Toncogene family member 2), ASXL1 (additional sex combs-like 1), CBL (casitas B-lineage lymphomaproto-oncogene), IDH1/IDH2 isocitrate dehydrogenase 1/2, IKZF1 (IKAROS family zinc finger 1), DNMT3A (DNA methyltransferase 3A), SOCS (suppressor of cytokine signaling), EZH2 (enhancer of zeste homolog2), TP53 (tumor protein p53), RUNX1 (runt-related transcription factor 1) ve HMGA2 (high mobility groupAT-hook 2) gibi birçok farklı mutasyon tanımlanmış (Tablo 2)24 ve MPN patogenez ve fenotipinde etkili olduğuna yönelik çalışmalar devam etmektedir.24,26
Tablo 2.Hematolojik malignitelerde edinilen gen mutasyonlarının frekans ve dağılımı.
Kırmızı ile boyalı alanlar belirtilen mutasyonların farklı hastalıklardaki frekansını gösterir. ALL =akut lenfositik lösemi; AML =akut myeloid lşösemi; CML= kronik myeloid lösemi; CNL=kronik nötrofilik lösemi; ET=esansiyal trombositoz; MDS=miyelodisplastik sendrom;RARS-T= Ring sideroblast ve trombositozla giden refrakter anemi; sAML=sekonder AML.24
7
2.1.1 Polisitemia Vera
ritropoezi düzenleyen mekanizmalardan bağımsız olarak eritrosit üretiminde artış ile kendini gösteren kemik iliğinin hipersellüler olduğu, her üç seride de hiperplazinin bulunduğu kronik miyeloproliferatif bir neoplazidir. PV, eritroid hücre artışı, lökositoz, trombositoz ve splenomegali ile karakterizedir ve ilk defa 1892’de Vaguez tarafından tanımlanmıştır.27
PV MPN’ler içinde en sık görülenidir ve primer polisiteminin en yaygın nedenidir. İnsidansı 2.3-2.8/100.000 olarak bildirilmiştir, ancak asemptomatik hastaların sıklığı nedeniyle hastalığın insidansının daha yüksek olduğu düşünülmektedir.Erkeklerde kadınlara göre daha sık görülmektedir.145
Ortalama tanı yaşı 60’tır ve nadiren 30 yaş altı hastalarda da görülebilir. Yapılan bir çalışmada hastaların %5’nin 0 yaş altında %1’nin ise 25 yaş altında tanı aldığı saptanmıştır.30-33
PV üç evrede tanımlanabilir;
1-Pre-polisitemik evre; hafif eritrositoz sınırında ve genelde belirgin trombositoz 2-Polisitemik evre
3-Post-polisitemik evre; anemiyi içeren sitopeni ve kemik iliği fibrozu, ekstramedüller hematopoezle karakterize edilir.62 astalar genellikle polisitemik dönemde tanı alırlar.
PV için tanı kriterleri ilk olarak 1960 yılı sonlarında PVSG (Polisitemia Vera Çalışma Grubu) tarafından düzenlenmiştir ve bu tanı kriterlerinde eritrosit kitle ölçümü zorunlu kabul edilmiştir. 2001 yılında DSÖ tarafından düzenlenen kriterlerde eritrosit kitle ölçümü yine yer almasına rağmen, zorunluluk çıkarılmış ve yüksek hemoglobin kriter olarak kullanılmıştır. 2008 yılında ise, JA 2 mutasyonun hemen her PV’lı hastada bulunması nedeni ile DSÖ tarafından revize edilmiştir ve günümüzde ilk önerilen tanı sistemidir. Buna göre, tanı için 2 ma or ve 1 minör kriter veya 1 ma or ve 2 minör kriter gereklidir.34 2013 yılında CALR mutasyonu tanımlandıktan sonra sıklığı, hastalığın seyri ve prognoza etkisi nedeniyle CALR ekzon 9 mutasyonlarının MP tanı algoritmasında olması gerektiği ilk defa 201 yılında önerildi.20
2015 yılı Ağustos ayından itibaren Dünya Sağlık Örgütü (WHO) PV, T ve PMF tanı kriterlerini güncelleyerek CALR mutasyonlarına yer verdi (Tablo 3,6,). CALR mutasyonu PV'da bulunmamaktadır.48
Sonuç olarak CALR mutasyonu, PV'da dışlama kriteridir.
Serum eritropoetin (EPO) seviyesi primer ve sekonder polisitemilerin ayrımında kullanılan bir test olup, PV’da düşük, sekonder polisitemilerde yüksektir. JA 2 V617F mutasyonu oldukça sensitif olup (sensitivite %97), düşük serum PO birlikteliği ile beraber spesifite %100’ dür. Düşük PO seviyesinin yanısıra JA 2 V617F mutasyonu negatif ise (PV’lı hastalarda %3’ü) ekson 12 JA 2 mutasyonu bakılmalıdır.35
8
Tablo 3. 2001-2015 yılları arasında WHO MPN tanı kriterlerinin evrimi20
ritrositler tipik olarak normokrom ve normositerdir, retikülosit artmaz. Lökosit sayısında belirgin artış görülebilir. Periferik yaymada miyelosit, metamiyelosit saptanabilir. Bazofil, eozinofil ve monosit sayısı tüm miyeloproliferatif hastalıklardaki gibi artar. Trombosit sayısı genellikle yüksektir. anama zamanı ve rutin koagülasyon testleri normaldir. Fakat hematokrit yüksekliğine bağlı olarak tüpteki plazma azaldığı için hatalı olarak PT ve aPTT yüksek bulunabilir.
W O’ya bağlı Myeloproliferatif Neoplaziler Araştırma ve Tedavi Uluslararası Çalışma Grubu (IWG-MRT) için tarafından 15 5 hastanın analizinden değerlendirilen en sık saptanan semptom ve bulgular; hemoglobin ~18. g/dl (15.1-26.5 g/dl), hematokrit ~%55 (%36-78), lökosit sayısı ~10. 00/μL (3.000-172.000/μL), trombosit sayısı ~466.000/μL (70.000-2.370.000/μL), LD yüksekliği %50, hipertansiyon % 6, splenomegali %36, kaşıntı %36, vazomotor semptomlar (eritromelal i gibi) %29, arteriyal tromboz %16, venöz tromboz %7, ma or hemora i % sıklığında saptanmıştır.47
Tromboz sık rastlanılan bir olaydır. Tedavi edilmemiş hastalar trombotik ve hemora ik olaylar açısından yüksek risklidir. İtalyan Polisitemia Vera Grubu verilerine göre hastaların %19’unda tromboz öyküsü vardır. Ölümcül olmayan tromboz ataklarının % 50’si venöz, %38’u arteriyeldir. Ölümle neticelenen trombozların %80’den fazlası
B1)WBC> 12X10*9/L EVET/HAYIR B2)KEMİK İLİĞİ HİSTOLOJİSİ PANMYELOZİS ÖN PLANDA ERİTROİD VE MEGAKARYOSİTİK PROLİFERASYON A2)SEKONDER ERİTROSİTOZ YOK
A3)SPLENOMEGALİ EVET/HAYIR
A4)KLONAL GENETİK ANOMALİ EVET/HAYIR A5)TROMBOSİTOZ >400X10*9 /L EVET/HAYIR
A2)JAK2 V617F VEYA
JAK2 EXON 12 MUTASYONU EVET
B2)NORMALİN ALTINDA
SERUM EPO DÜZEYİ EVET/HAYIR B3)ENDOJEN ERİTROİD
KOLONİ FORMASYONU EVET/HAYIR
A3)JAK2 MUTASYONU EVET
Tablo 3. 2001-2015 yılları arasında MPN Polisitemia Vera WHO tanı kriterlerinin evrimi
ERKEK>18,5 KADIN>16,5 VEYA ARTMIŞ PANMYELOZİS (YAŞA GÖRE DÜZELTİLMİŞ HİPERSELLÜLARİTE VE HER ÜÇ SERİDE ARTIŞ) B1)KEMİK İLİĞİ HİSTOLOJİSİ
2008 WHO TANI KRİTERLERİ (2 MAJOR VE 1 MİNÖR KRİTER YA DA A1 VE 2 MİNÖR KRİTER)
A1)HEMOGLOBİN (g/dl) KIRMIZI HÜCRE KÜTLESİ HEMATOKRİT (%)
A1)HEMOGLOBİN (g/dl) KIRMIZI HÜCRE KÜTLESİ HEMATOKRİT (%)
ERKEK>18,5 KADIN>16,5 VEYA ARTMIŞ 2001 WHO TANI KRİTERLERİ
(A1+A2 İLE TOPLAM 3 MAJOR KRİTER YA DA A1+A2 VE 2 MİNÖR KRİTER)
MAJOR KRİTERLER MİNÖR KRİTERLER
A1)HEMOGLOBİN (g/dl) KIRMIZI HÜCRE KÜTLESİ HEMATOKRİT (%) ERKEK>16,5 KADIN>16,0 VEYA ARTMIŞ ERKEK>%49 KADIN>%48 B1)NORMALİN ALTINDA
SERUM EPO DÜZEYİ EVET/HAYIR
PANMYELOZİS (YAŞA GÖRE DÜZELTİLMİŞ HİPERSELLÜLARİTE,HER ÜÇ
SERİDE ARTIŞ VE PLEOMORFİK MATÜR
MEGAKARYOSİTLER) A2)KEMİK İLİĞİ HİSTOLOJİSİ
2015 WHO TANI KRİTERLERİ (3 MAJOR YA DA A1+A2 VE MİNÖR
9 arteriyel olduğu görülmüştür.36 İskemik inme, miyokard infarktüsü ve geçici iskemik
atak en yaygın trombotik olaylardır. Hastalarda derin ven trombozu, pulmoner emboli, periferik vaskuler oklüzyon da sık görülmektedir. PV ile ilişkilendirilmiş ciddi bir trombotik olay da hepatik venöz veya inferior vena cava trombozunun neden olduğu Budd-Chiari sendromudur.37-42 Bunlar dışında sık saptanan fizik muayene bulguları ise fasial pletore, olguların bir kısmında ekstramedüller hematopoeze bağlı hepatomegali, hiperürisemiye bağlı gut artriti ve tofüstür.
Tedavi risk grubuna göre belirlenir. Çok yüksek trombosit sayısı (>1.500.000/μL) kanama açısından potansiyel risk faktörüdür. Lökosit sayısı ve yüksek JA 2 V617F allel yükü tromboz açısından potansiyel risk faktörleridir. PV risk sınıflaması aşağıdaki tabloda verilmiştir. (Tablo )49,50
Tablo . Polisitemia vera olgularında risk sınıflaması
Risk Kategorisi Yaş >60 veya tromboz öyküsü var Genel kardiyovasküler risk faktörleri* Düşük Yok Yok
Orta Yok Var
Yüksek Var Var veya yok
*Hipertansiyon, hiperkolesterolemi, diyabet, sigara içimi
Tablo 4. Polisitemia vera olgularında risk sınıflaması
Düşük riskli olan hastalarda tedavi algoritması flebotomi ve düşük doz aspirin (80mg/g) şeklindedir. Aspirin hemora i öyküsü olan veya edinilmis von willebrand sendromu veya çok yüksek sayılarda trombositozu olan olgularda artmış kanama riski nedeniyle kullanılmamalıdır. Yüksek riskli olan hastalarda ise flebotomi, aspirin ve miyelosupresif tedavi (hidroksiüre, busulfan, anagrelid, interferon alfa, radyoaktif fosfor) şeklindedir.51
Flebotomi için hedef hematokrit seviyeleri kadınlarda % 2, erkeklerde % 5’de tutulmalıdır. aftada iki kez ya da daha küçük volümlerle flebotomi yapılması önerilmektedir.51
10 Tablo 5.PV tedavi yönetimi
* Anagrelide trombositoz nedeniyle tedavi gereksinimi olan ve ilk seçenek tedavileri tolere edemeyen veya ilk seçenek tedavilerle trombositozu kontrol edilemeyen hastalarda kullanılır52
Akut lösemiye dönüşüm olasılığı düşüktür ancak bazı hastalar önceki sitotoksik terapilerle ilişkili olarak miyelodisplastik ya da blastik evre geliştirebilir. Tedavisiz ortalama yaşam süresi tanı sonrası 6-18 ay olarak tahmin edilirken tedavi ile hastalarda bu süre 10 yıldan fazladır.28,29
PV’li hastalar %5 ile %50 oranında tanı aldıktan ortalama 10 yıl sonra MF’ye dönüşüm riski vardır. Post-PV MF’si mevcut olan PV’li hastaların akut lösemiye dönüşme riski daha yüksektir. Son evreye giren hastaların %20-50’sinde lösemik transformasyon gözlenir.43,44
Ruxolitinib ABD’de FDA tarafından miyelofibrozis ve post-polisitemik miyelofibroz tedavisinde kullanımı onaylanan bir JAK1, JAK2, JAK3 ve TYK2 inhibitörüdür. İlacın JA 2, STAT5, R 1/2 ve Akt fosforilasyonunu baskıladığı gösterilmiştir. Ruxolitinibin ayrıca PV’li hastalarda endo enlerin ve sitokin-destekli eritroid progenitörlerin gelişimini de inhibe ettiği gösterilmiştir.53
2.1.2 Esansiyel Trombositoz
Esansiyel trombositemi etyolojisi bilinmeyen, pluripotent hematopoietik kök hücredeki bozukluk nedeni ile oluşan, klinik olarak açıklanabilir bir neden olmaksızın trombosit sayısındaki belirgin artış ile karakterize hematolo ik klonal bir hastalıktır.54
sansiyel trombositemi insidansı 100.000’de 2,5-10 arasındadır. adınlarda erkeklere göre 2 kat fazla görülür.146 Genellikle asemptomatik olması nedeniyle gerçek görülme
sıklığının daha fazla olduğu düşünülmektedir.25 Görülme yaşı ortalama 60 yaştır fakat 0
yaş altında da yaklaşık % 20 görülürken çocukluk çağlarında nadirdir.55
astaların 2/3’ü asemptomatik olmakla birlikte klinikte splenomegali, tromboz ve hemora ilerle seyreden kronik bir MP ’tır.
İlk defa 1934 yılında tekrarlayan hemora ik epizodları olan ve platelet sayısı artmış bir hastada Epstein ve Goedel tarafından tanımlanmıştır. 2005 yılında JA 2
YAŞ 1. BASAMAK TEDAVİ 2. BASAMAK TEDAVİ 3. BASAMAK TEDAVİ 4. BASAMAK TEDAVİ
<60 IFN-α / HU HU Anagrelide*
60-75 HU IFN-α Anagrelide*
>75 HU Kombine Tedavi Busulfan P32 RADYOAKTİF
Tablo 5. PV Tedavi Yönetimi
11 mutasyonunun keşfinden sonra hastalığın patogenezi, tanısı ve sınıflamasında önemli değişiklikler ortaya çıkmıştır.
sansiyel trombositemi tanısı reaktif trombositoz ve kronik miyeloid bozuklukların varlığının dışlanması ile konulur .56 Tanı kriterleri oluşturulurken de bu
özellik dikkate alınarak diğer sebeplerin dışlanması da kriterler içerisinde yerini almıştır. 2005 yılından beri T patogenezinde de JA 2, MPL ve CALR mutasyonlarının sıklığı ve fenotipe etkisi nedeniyle 2015 için önerilen W O kriterleri olarak tanı kriterlerine eklenmiştir.20
(Tablo 6)
Tablo 6. 2001-2015 yılları arasında MPN Esansiyel Trombositemi WHO tanı
kriterlerinin evrimi20
T hastalarının yarısı asemptomatik olup rutin kan tetkiklerinde saptanan trombositoz ile tanı alırlar. Semptomatik olanlarda sıklıkla baş ağrısı, baş dönmesi, senkop, atipik göğüs ağrısı, akral paresteziler, livedo retikülaris, eritromelal i ve görme bozuklukları gibi vazomotor semptomlar ve trombohemora ik komplikasyonlar görülür Ancak T için spesifik bir semptom veya bulgu yoktur.57
Mikrovasküler belirtilerin patogenezinde artmış tromboksan sentezi ön plandadır ve aspirin tedavisi ile önlenebilir.58
Tromboz oluşumunda lökositlerin de rolü vardır. Lökositoz tromboz için bağımsız bir risk faktörüdür.59
Hemorajik komplikasyonlar edinsel von Willebrand hastalığı ile ilişkilidir. Bu durum trombosit sayısının artması ve
A2)KEMİK İLİĞİ HİSTOLOJİSİ
BÜYÜK, OLGUN MEGAKARYOSİTLERDE SAYI ARTIŞI İLE GİDEN ÖZELLİKLE MEGAKARYOSİTER SERİDE ARTIŞ
A1)TROMBOSİT SAYISI ≥450X10*9/L
A2)KEMİK İLİĞİ HİSTOLOJİSİ
BÜYÜK, OLGUN MEGAKARYOSİTLERDE SAYI ARTIŞI İLE GİDEN ÖZELLİKLE MEGAKARYOSİTER SERİDE ARTIŞ, NÖTROFİLLERDE SOLA KAYMA YA DA ERİTROPOEZDE, GRANÜLOPOEZDE ARTIŞ OLMAMASI
A3) PV, KML, PMF, MDS VEYA DİĞER MYELOİD NEOPLAZMLARA
AİT HERHANGİ BİR VERİ OLMAMASI A4) JAK2 V617F VEYA DİĞER KLONAL MARKERLARIN OLMASI VE REAKTİF TROMBOSİTOZA AİT VERİ OLMAMASI A1)TROMBOSİT SAYISI
≥450X10*9/L
A3) PV, KML, PMF, MDS VEYA DİĞER MYELOİD NEOPLAZMLARA
AİT HERHANGİ BİR VERİ OLMAMASI A4)JAK2, CALR VEYA MPL MUTASYONU
B1)KLONAL MARKER VARLIĞI YA DA REAKTİF TROMBOSİTOZA AİT VERİ OLMAMASI
BÜYÜK, OLGUN MEGAKARYOSİTLERDE SAYI ARTIŞI İLE GİDEN ÖZELLİKLE MEGAKARYOSİTER SERİDE ARTIŞ, NÖTROFİLLERDE SOLA KAYMA YA DA ERİTROPOEZDE, GRANÜLOPOEZDE ARTIŞ OLMAMASI, GRADE 1 RETİKÜLİN FİBERDE ARTIŞ OLMASI
A2)KEMİK İLİĞİ HİSTOLOJİSİ ÖNERİLEN 2015 WHO TANI
KRİTERLERİ
(4 MAJOR YA DA 3 MAJOR VE 1 MİNÖR KRİTER)
Tablo 6. 2001-2015 yılları arasında MPN Esansiyel Trombositemi WHO tanı kriterlerinin evrimi
2008 WHO TANI KRİTERLERİ (4 MAJOR KRİTER)
A1)TROMBOSİT SAYISI ≥600X10*9/L
2001 WHO TANI KRİTERLERİ (3 MAJOR KRİTER)
MAJOR KRİTERLER MİNÖR KRİTERLER
A3) PV, KML, PMF, MDS VEYA REAKTİF TROMBOSİTOZA AİT HERHANGİ BİR VERİ OLMAMASI
12 yüksek moleküler ağırlıklı vWF’ünün trombositlerle etkileşmesi ve proteolizisine bağlıdır.60
astalarda görülen epinefrin-ADP-kollajen trombosit agregasyonunun bozulması, azalmış ADP sekresyonu ve trombosit depo kusurları gibi diğer trombosit kusurları kanama riskini az da olsa etkiler.61
ET’nin en önemli fizik muayene bulgusu hastaların %25- 8’inde gözlenebilen splenomegalidir.70 epatomegali ve lenfadenopati nadir bulgulardır. Parmak uçlarında renk değişikliği, eritromelal i ve gangrenler sık görülen cilt bulgularıdır.
Trombosit sayısı hastaların tümünde 50.000/mm3’den, çoğunda 1.000.000/ mm3 fazladır. emoglobin genellikle normaldir. Anemi görülebilir. Nötrofilik lökositoz görülür. Lökosit formülünde sola kayma, eozinofili ve bazofili sıklıkla görülür. Dev trombositler gözlenebilir.71 Lökosit alkalen fosfataz (LAP) skoru normal veya yüksektir.
LD ve ürik asit yüksek olma eğilimindedir. Belirgin trombositozu olan olgularda psödohiperkalemi görülebilir. Protrombin ve parsiyel tromboplastin zamanı normaldir, ancak uzamış kanama zamanı ve bozulmuş trombosit agregasyonu gibi trombosit fonksiyonu anormallikleri görülebilir.72
emik iliği hiperselülerdir ve belirgin megakaryosit artışı dikkat çekicidir. Artmış ploidiye sahip dev megakaryositler kümeleşmiş olarak görülür. Sıklıkla eritroid ve granülositer dizi hiperplaziye eşlik eder. afif düzeyde fibrozis görülebilir. Fibrozisin belirgin olması T aleyhine bir bulgudur, PMF’nin erken evrelerini düşündürür. Demir skoru yüksektir.73
X-bağımlı genlerin polimeraz zincir reaksiyonu ve DNA metilasyonundan yararlanılan çalışmalar sonrası T’li hastaların büyük çoğunluğunun monoklonal hematopoeze sahip olduğu görülmüştür. JA 2V617F mutasyonu vakaların yaklaşık %50’sinde, MPLW515K/L mutasyonu ise %1- ’ünde tanımlanmıştır.63,64
Bu mutasyonlar megakaryosit proliferasyonu ve trombosit üretimini uyaran yolakların aşırı aktivasyonuna neden olmaktadır.65
2013 yılında tanımlanan CALR mutasyonu ise bu hastaların %15-25’inde tanımlanmıştır.63,64
n büyük megakaryositik gelişme ve farklılaşma faktörü olan serum trombopoetin seviyesinin normal ya da yükselmiş olduğu gösterilmiştir. Trombopoetinin sirkülasyon seviyesi, MPL’ye bağlanma derecesiyle düzenlenmektedir. Megakaryositlerin ve trombositlerin kütlesi artarken, bu bağlanmanın sonucu olarak trombopoetin seviyesi düşer. Normal olarak MPL-trombopoetin kompleksi yok edildiği kadar trombositler sirkülasyondan uzaklaştırılır, trombopoetin seviyeleri daha fazla trombosit üretimini uyarmak için artar. Megakaryositlerde trombopoetinin MPL ile bağlanması normalde MPL’nin konformasyonel olarak değişimini ve JA kinazın MPL’nin sitoplazmik
13 domainine aşırı derecede bağlanmasını başlatır. Bu, STAT, PI3 ve MAP yolakları aracılığıyla proliferasyon, endoreduplikasyon ve megakaryosit kütle artışı uyaran sinyalizasyonu başlatır.66
astaların % 5’inden azında akut lösemiye transformasyon görülmektedir. Ortalama sağkalım süresi 10-15 yıl olarak belirtilmiştir. omplikasyonların kontrole alındığı durumlarda normal yaşam süresine sahiptirler.74
Mortalite lösemi veya miyelofibroza dönüşüm ve trombotik ya da hemora ik komplikasyonlara bağlıdır. emoglobin düzeyinin normalden az olması, 60 yaş üstünde olma, Lökosit sayısı 15.000/mm³ üstünde olması tanımlanmış risk faktörleridir. Benzer biçimde anemi ve aşırı yüksek trombositoz (1.500.000/mm³) birlikteliği lösemik dönüşüm riskini artırır. Mevcut tedavi yöntemleri ne lösemiye ne de miyelofibroza dönüşümünü azaltmaz, sağ kalıma da belirgin bir etkisi yoktur. Bu nedenle agresif tedavi gereksizdir, düşük riskli hastalarda ilk basamak tedavi seçeneği aspirindir. Amaç komplikasyonların önlenmesi ve yönetimidir. ğer aspirinle şikayetler azalmıyorsa veya hastanın risk grubu orta/yüksekse sitoredüktif tedavi önerilir.
Sitoredüktif tedavide ilk seçenek hidroksiüredir. idroksiüre ile birlikte aspirin kullanımı, mikrovasküler şikâyetleri rahatlatır ve tromboz ve kanama ihtimalini azaltır.67
Yüksek riskli hastalarda trombosit sayısını 00.000/mm³’nin altına düşürülmesi hedeflenir. ğer hidroksiüre semptomları veya trombositozu yeterince kontrol edemiyor veya yan etkileri nedeniyle kullanılamıyorsa anagrelid kullanılabilir.68
Hamile veya doğurganlık çağındaki hastalarda interferon alfa kullanılabilir. Alkilleyici ilaçlar (busulfan, klorambusil) lösemiye neden olan yan etkileri nedeniyle kullanılmamaktadır, çok yaşlı ve beklenen ömrü az olan hastalarda kullanılabilir. JA 2 inhibitörleri de T tedavisinde deneysel amaçlı kullanılmaktadırlar.69
2.1.3 Primer Miyelofibroz
W O tarafından kronik idiopatik miyelofibrozis olarak adlandırılan primer miyelofibrozis (eski adı ile agno enik miyeloid metaplazi) miyeloid hücrelerin değişken morfolojik maturasyonu ve klonal proliferasyonu ile karakterize bir myeloproliferatif neoplazidir.75,76 emik iliğinin fibrozisi, ekstrameduller hematopoez ile birlikte miyeloid metaplazi ve splenomegali ile karakterize nadir görülen bir hastalıktır. Yıllık insidansı yaklaşık 100.000’de 1,5’tur. Genellikle 60 yaş üzerinde görülmektedir. rkeklerde daha sık görülür.76,77
İyonize radyasyon, benzen ve hidrokarbonlara maruziyetin PMF nedeni olabileceği ileri sürülse de etyolo isi net olarak belli değildir.
14 20q- ve 13q- kromozom delesyonları başta olmak üzere del(6)t(1;6) (q21-23;p21.3), 9p, trizomi 8 veya 9, - 18 - kısmi trizomi 1q gibi spesifik olmayan kromozom anomalileri yaygın olarak görülür.76
Anöploidi veya psödoploidi sıktır. JA 2 mutasyonu PMF’li hastaların %50’sinde pozitif bulunur.78
Sıklıkla homozigottur. MPLW515L/ olguların %5’inde , CALR Tip 1 (52-bp delesyon) ve Tip 2 (5-bp insersiyonu) ise %20-35’inde pozitif saptanmıştır.79,10,13
astalık giderek artan anemi, kaşeksi ve splenomegali ile seyreder. PMF’de hemen her organda ekstramedüller hematopoez odakları gelişebilir. astaların çoğu eritrosit transfüzyonuna bağımlıdır. Az bir kısmında semptomatik trombositopeni ve nötropeni vardır. Progresif splenomegali ve lökositoz ile hastalarda kaşeksi, periferik ödem, belirgin halsizlik, yorgunluk, gece terlemesi, kabızlık ve düşük derecede ateş görülmesi arasında ilişki vardır. Bazı hastalarda semptomatik portal hipertansiyon gelişir, özofagus varisleri ve varis kanamaları görülür. Üst ve alt ekstremite ağrıları ve non-trombotik pulmoner hipertansiyon görülebilir. Bunun yanında özellikle trombositozlu hastalarda kanama veya tromboz komplikasyonları oluşabilir.
Periferik yaymanın en önemli özelliği miyelofitizidir. Bu durum kendisini çekirdekli eritrosit seri hücreleri, metamiyelosit, miyelosit, miyeloblast ve megakaryosit varlığı (lökoeritroblastozis) ile gösterir. astaların 2/3’ünde anemi vardır. Lökositoz %41- 9, lökopeni %7-22, trombositoz %13-31, trombositopeni %21-37, periferik kanda blast %33-53, LD artışı ise %83 oranında gözlenir.74
PMF tanısında fibrozisin gösterilmesi ve malignitenin dışlanması için kemik iliği biyopsisi yapılmalıdır. Biyopsi ile kemik iliğindeki fibrozis gösterildikten sonra PMF tanısının doğrulanması için kemik iliği fibrozisine yol açabilecek kronik miyeloproliferatif hastalıklar, miyelodisplastik sendrom, akut lösemiler, lenfoid hastalıklar, kemik iliği metastazı yapmış solid tümörler, bağ dokusu hastalıkları, infeksiyonlar ve D vitamini eksikliği gibi diğer nedenler dışlanmalıdır. emik iliği genellikle aspire edilemez (Dry tap). Biyopside fibrozisin belirgin olmadığı hipersellüler bir ilikten, tamamıyla fibrotik hatta osteosklerotik iliğe varan tablo görülebilir. Megakaryositler sayıca artmış ve displazik görünümdedir. Granülositler hiper veya hipolobülasyon, edinsel Pelger- uet anomalisi ve nükleositoplazmik asenkroni görülebilir. arakteristik bir bulgu, dilate sinüsler içinde immatür hücre gruplarının bulunmasıdır. 82
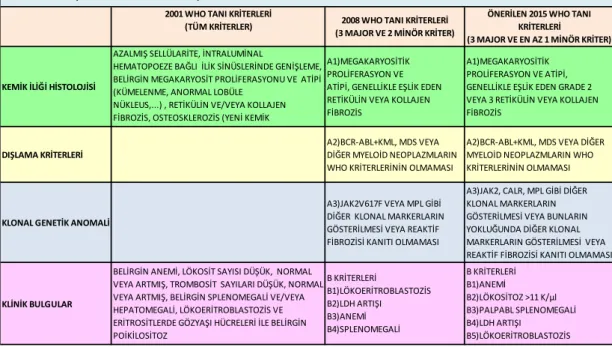
15 2005 yılından beri PMF patogenezinde de JA 2, MPL ve CALR mutasyonlarının sıklığı ve fenotipe etkisi nedeniyle 2015 için önerilen W O kriterleri olarak tanı kriterlerine eklenmiştir (Tablo 7).20
Tablo 7. 2001-2015 yılları arasında MPN Primer Miyelofibrozis WHO tanı kriterlerinin
evrimi (***JAK2,CALR, MPL mutasyon yokluğunda en sık eşlik eden mutasyonlar ASXL1, EZH2, TET2, IDH1/IDH2, SRSF2, SF3B1 hastalığın klonal doğasını belirlemede yardımcı olacaktır.)20
Semptomatik anemi, splenomegali, karaciğer ve dalak dışındaki ekstramedüller hematopoez, kemik ağrısı, ekstramedüller hematopoez ilişkili pulmoner hipertansiyon veya konstitüsyonel semptomların (örneğin halsizlik, gece terlemesi ve pruritis) varlığında tedavi başlanması gerekmektedir.83
Belirgin lökositoz veya trombositozu olan hastalarda sitoredüktif tedavi başlanabilir. Anemi varlığında andro enler, steroid veya lenalidomid ile tedavi planlanabilir.84,85 Del(5q31) varlığında lenalidomid etkilidir.86 Anemisi olup splenomegalisi olmayan hastalarda eritropoetin tedavisi faydalı olabilir. Anemisi olan hastaların serum PO düzeyinin 125 U/L nin altında olması ve transfüzyon bağımlı olmaması durumunda eritropoetin tedavisinden fayda gördükleri saptanmıştır.83
Splenomegalisi olan hastalarda ilk basamak tedavi hidroksiüre olup hastaların yaklaşık % 0’ında dalak boyutu hidroksiüre ile yarı yarıya azalmaktadır.87
Talidomid ve lenalidomidin splenomegaliyi gerilettiği ve trombositopeniyi düzelttiği gösterilmiştir.88
JA 2 inhibitörleri miyelofibrozis tedavisinde splenomegaliyi ve konstitüsyonel semptomları azaltmada etkilidir. Bu etkisi dramatiktir, fakat ilaç kesilince sitokin
KEMİK İLİĞİ HİSTOLOJİSİ
DIŞLAMA KRİTERLERİ
KLONAL GENETİK ANOMALİ
KLİNİK BULGULAR
A2)BCR-ABL+KML, MDS VEYA DİĞER MYELOİD NEOPLAZMLARIN WHO KRİTERLERİNİN OLMAMASI A3)JAK2, CALR, MPL GİBİ DİĞER KLONAL MARKERLARIN GÖSTERİLMESİ VEYA BUNLARIN YOKLUĞUNDA DİĞER KLONAL MARKERLARIN GÖSTERİLMESİ VEYA REAKTİF FİBROZİSİ KANITI OLMAMASI B KRİTERLERİ B1)ANEMİ B2)LÖKOSİTOZ >11 K/μl B3)PALPABL SPLENOMEGALİ B4)LDH ARTIŞI B5)LÖKOERİTROBLASTOZİS
Tablo 7. 2001-2015 yılları arasında MPN Primer Miyelofibrozis WHO tanı kriterlerinin evrimi
BELİRGİN ANEMİ, LÖKOSİT SAYISI DÜŞÜK, NORMAL VEYA ARTMIŞ, TROMBOSİT SAYILARI DÜŞÜK, NORMAL VEYA ARTMIŞ, BELİRGİN SPLENOMEGALİ VE/VEYA HEPATOMEGALİ, LÖKOERİTROBLASTOZİS VE ERİTROSİTLERDE GÖZYAŞI HÜCRELERİ İLE BELİRGİN POİKİLOSİTOZ
A1)MEGAKARYOSİTİK PROLİFERASYON VE ATİPİ, GENELLİKLE EŞLİK EDEN RETİKÜLİN VEYA KOLLAJEN FİBROZİS
A2)BCR-ABL+KML, MDS VEYA DİĞER MYELOİD NEOPLAZMLARIN WHO KRİTERLERİNİN OLMAMASI
A3)JAK2V617F VEYA MPL GİBİ DİĞER KLONAL MARKERLARIN GÖSTERİLMESİ VEYA REAKTİF FİBROZİSİ KANITI OLMAMASI
B KRİTERLERİ
B1)LÖKOERİTROBLASTOZİS B2)LDH ARTIŞI B3)ANEMİ B4)SPLENOMEGALİ
2001 WHO TANI KRİTERLERİ
(TÜM KRİTERLER) 2008 WHO TANI KRİTERLERİ (3 MAJOR VE 2 MİNÖR KRİTER)
ÖNERİLEN 2015 WHO TANI KRİTERLERİ
(3 MAJOR VE EN AZ 1 MİNÖR KRİTER)
AZALMIŞ SELLÜLARİTE, İNTRALUMİNAL
HEMATOPOEZE BAĞLI İLİK SİNÜSLERİNDE GENİŞLEME, BELİRGİN MEGAKARYOSİT PROLİFERASYONU VE ATİPİ (KÜMELENME, ANORMAL LOBÜLE
NÜKLEUS,...) , RETİKÜLİN VE/VEYA KOLLAJEN FİBROZİS, OSTEOSKLEROZİS (YENİ KEMİK FORMASYONU)
A1)MEGAKARYOSİTİK PROLİFERASYON VE ATİPİ, GENELLİKLE EŞLİK EDEN GRADE 2 VEYA 3 RETİKÜLİN VEYA KOLLAJEN FİBROZİS
16 fırtınası nedeniyle şikayetleri aniden artar. Trombositopeni ve anemi gibi yan etkileri vardır.90
Düşük riskli ve 5 yaşın altındaki genç hastalarda uygun verici varsa allojenik kök hücre nakli yapılabilir. 65 yaşın altında ise düşük yoğunluklu tedavi ile allojenik nakilden fayda görebilir.89
Ortalama yaşam süresi 5 yıldır. Diğer MPN’lere göre yaşam süresi daha kısa ve semptomların görülme sıklığı ve çeşitliliği fazladır. Başta akciğer infeksiyonları olmak üzere infeksiyonlara yatkınlık mevcuttur. PMF’de blastik dönüşüm ilk on yılda %2-16 oranında gözlenir. Blastik dönüşüm olan hastalarda mortalite çok yüksektir, agresif tedavi destek tedavisinden daha iyi değildir. Diğer sık karşılaşılan ölüm nedenleri %26-29 enfeksiyon, %11-22 kanama, %7-15 kalp yetmezliği, %3-8 karaciğer yetersizliği, %3 solid tümör, %3 solunum yetersizliği ve %6 portal hipertansiyondur.80,81
2.2 KRONİK MİYELOPROLİFERATİF HASTALIKLARDA TANIMLANMIŞ MUTASYONLAR
JAK2 Mutasyonları
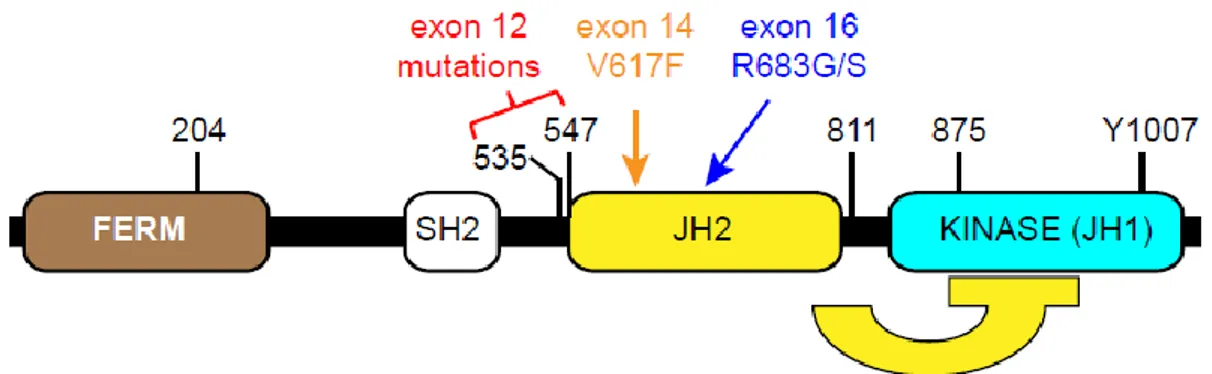
Janus kinaz (JAK) ailesi, JAK-STAT yolağı ile, sitokin aracılıklı sinyallerin dönüşümünü sağlayan bir grup (hücre içi reseptör olmayan) tirozin kinaza verilen isimdir. Bu yolaktaki transkripsiyon faktörleri ise STAT’lar (Signal Transducers and Activators of Transcription) olarak bilinir. 9p2 ’te yer alan JA 2 geninin 25 ekzonu vardır. 1132 aminoasitten oluşan bir protein kodlamaktadır.91
Her JAK bir aktif tirozin kinaz domaini JA homolo i 1 (J 1), bir katalitik olarak inaktif psödokinaz domaini JAK homoloji 2 (JH2), bir SRC homoloji 2 domaini (SH2) ve tip 1 sitokin reseptörlerinin bağlandığı bir amino terminal F RM homolo i domaini içermektedir.92
17 JA ailesi, JA 1, JA 2, JA 3 ve Tirozin kinaz 2 (TY 2) olmak üzere dört üyeden oluşur. JAK1 ve JAK2 tip II interferon (IFN-γ) sinyal yolunda rol alırken, JAK1 ve TYK2 tip I IFN sinyallenmesi ile ilişkilidir.94 TY 2 “natural killer” fonksiyonlarına aracılık eder.95
JA 2 eritropoetin, trombopoetin, interlökin-3, granülosit stimüle edici faktör (G-CSF), ve granülosit-makrofa stimüle edici faktör (GM-CSF) reseptörleri üzerinden intraselüler sinyal iletiminde rol oynar. Reseptöre bağlanma sonrasında fosforillenmesi ve aktive olması ile reseptörde yapısal değişikliğe sebep olur. Aktive JA 2 reseptörünün sitozolik parçasını fosforilleyerek intraselüler sinyal iletimini başlatarak hücre proliferasyonunu uyarır.93
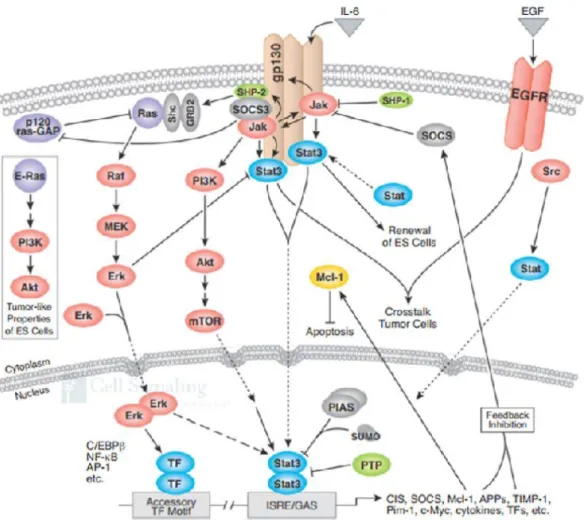
JAK-STAT yolunda her reseptör, kendi ligandı (sitokin) ile bağlandığında, her iki JAK domainini birbirlerini fosforile edecek şekilde yakınlaştıran bir konformasyonel değişime uğrar. Ligandın reseptöre bağlanması kinaz aktivitesinde artışa neden olur. Bu sayede tirozin rezidüleri fosforile olur ve reseptörde S 2 domain (fosfotirozin bağlanma bölgesi) bulunduran proteinlerle etkileşime girebilecek bölgeler ortaya çıkar. Bu fosfotirozin rezidülerine bağlanabilen, SH2 domaine sahip olan STAT’lar reseptörde birikir ve bunlar da JA ’lar tarafından tirozin-fosforilasyona uğrar. Farklı STAT’lar üzerine eklendikçe hetero ve homodimerler meydana gelir. Aktive olan dimerler hücre çekirdeğinde birikerek hedef genlerin transkripsiyonunu aktive eder. STAT’ların direkt olarak reseptör tirozin kinazları tarafından tirozin-fosforillenmesi (örneğin epidermal growth faktör reseptörü) ve hatta reseptör dışı tirozin kinazlarla fosforillenmesi (c-src,… gibi) de mümkündür.96
JAK-STAT yolağı birçok yolla negatif yönde regüle edilir. Bunlardan bir tanesi olan protein tirozin fosfatazlar, hem sitokin reseptörlerinden, hem de aktive STAT’lardan fosfatları ayırır.96
Bir başka mekanizma olan SOCS (Suppressors of Cytokine Signaling) sistemi JA ’ları bağlamak, ya da STAT’lar ile reseptör bağlanma bölgeleri için yarışarak STAT fosforilasyonunu inhibe eder.98
STAT’ların, PIAS (Protein Inhibitors of Activated STATs) isimli negatif regülatörlerle hücre çekirdeğinde inhibisyonu da mümkündür.97
Kısaca, JA otofosforilasyonu kendi içinde bir konformasyonel değişimi indükler ve daha ileri aşamalardaki transkripsiyon faktörlerini (STAT’lar) fosforile ve aktive eder. Aktive olan STAT’lar reseptörden ayrılır ve hücre çekirdeğine giderek seçilmiş genlerin transkripsiyonunu düzenler.96 (Şekil 2.)
18
Şekil 2. JAK-STAT Yolu. (www.cellsignal.com)26
JA 2V617F mutasyonunun keşfi Vainchenker ve arkadaşları tarafından gerçekleşmiş ve 2005’de yayınlanmıştır.8
JAK2V617F mutasyonu JA 2’nin J 2 parçasındaki 617. odonunda guanin timin değişiminden dolayı valinin fenilalanin ile yer değiştirmesi sonucu ortaya çıkar. J 2 domainindeki bu mutasyon J 1 kinaz domaini otoinhibisyonuna neden olur ve sonuçta JA ileti sistemindeki inhibisyon ortadan kalkar. Böylece her 3 dizinin de etkilendiği bir proliferasyon durumu ortaya çıkar. JA 2V617F mutasyonu ile PV hastalarının %95-98’inde, T ve PMF hastalarının %50’sinde ( T’de %50-70, PMF’de % 0 ) klonal gelişimi göstermek mümkün olmuştur.4-8,99
omozigot (+) olan PV hastalarında daha büyük splenomegali ve artmış kardiovasküler risk ile ilişkilendirilmiş.131
PMF hastalarında JA 2V617F mutasyonu varlığı splenomegalide, lökosit, hemoglobin ve trombosit sayılarında ve blastik dönüşüm riskinde artış ile ilişkili bulunmuştur.132
JAK2 (+) PMF ‘de artmış lökosit sayıları daha kötü sağkalım için anlamlı bulunmuştur.133
19 T’de JA 2V617F mutasyonunun varlığı b ve lökosit sayılarında artış, trombosit sayılarında daha düşük seyirle birlikte daha geç yaşta tanı alma ve venöz tromboz riskinde artış ile ilişkili bulunmuştur.134-136
JA 2V617F mutasyonu olan PV hastalarında b ve nötrofil sayıları yüksek seyretmekle birlikte bu durum yüksek trombotik olayla ilişkilendirilememiştir. Ancak JA 2 allel yükü ne kadar fazla ise miyelofibrozise transformasyon ve kaşıntı şikayetinde artış o kadar belirgindir.135,137,138
JAK2V617F mutasyonu %5 (-) olguda %30 oranında saptanan bir diğer somatik kazanılmış mutasyon JA 2 exon 12 mutasyonudur. Genellikle heterozigottur. SH2 ve J 2 domainleri arasında bağ görevi görür.100,101
Her ne kadar JH2 domaini üzerinde yer almasa da tıpkı JA 2V617F mutasyonu gibi etki gösterir. Ailesel olgularda gözlenmektedir. Bununla birlikte T ve PMF hastalarında gözlenmez ancak PV hastalarında miyelofibroza gidişle ilişkili bulunmuştur.102
MPL Mutasyonu
MPL (Miyeloproliferatif Lösemi Virüs Onko en) geni kromozom 1p3 ’de bulunur ve 12 ekzondan oluşur. MPL (trombopoetin reseptörü), proto-onkogen c-MPL tarafından kodlanır. Trombopoetin reseptörü c-MPL aracılığıyla megakaryopoez ve trombopoezin önemli bir regülatörünü temsil eden bir trombopoetindir. Bu reseptör, CD3 +
hematopoetik progenitörleri, megakaryositler ve trombositler üzerinde ifade edilir. MPL mutasyonu reseptörün ukstamembranöz bölgesinde oluşur. kzon 10’da 515. kodon üzerindeki triptofanın lösin, lizin, aspara in veya alanin ile yer değiştirmesi sonucu oluşur.103-105
Bu molekül reseptörün sitozolik şekli için önemli bir rol oynar ve spontan aktivasyonunu önler. Bu yer değiştirme mutasyonlarından sonra reseptörün inhibisyon mekanizması kaybolarak hücrede anormal proliferasyon olur.
MPL için tanımlanan mutasyonlardan W515 (Triptofan – Lizin) ve W515L (Triptofan - Lösin) en sık görülen mutasyonlardır. MPL mutasyonlarının MPN’de görülme sıklığı PMF ve T’de %3-8 şeklindedir.79,106
Pikman ve arkadaşlarının yaptığı çalışmada fareye yapılan kemik iliği transplantasyonu ile ortaya çıkan MPLW515L mutasyonu trombositoz, splenomegali, splenik infarkt ve miyelofibrozis ile ortaya çıkan MPN ile karakterizedir.139
MPL mutasyonları MPN’de yüksek trombosit ve serum eritropoetin seviyeleri ve düşük b , azalmış kemik iliği sellülaritesi ve daha genç yaşta prezentasyonun yanı sıra transfüzyon bağımlılığı ve trombotik komplikasyonlarda artış ile ilişkilidir.140-143
20
LNK Mutasyonu
Lenfosit-spesifik Adaptör Protein (LN ), bir JA -STAT inhibitör adaptör proteinidir. S 2B3, S 2B gen ailesinin bir üyesidir. Bu ailenin üyeleri 3 ana bölümden (prolinden zengin amino bölgesi, plekstrin homolo i ve S 2 domainleri) ve C bölgesindeki tirozinden meydana gelir. LNK hematopoezde JAK2 aktivasyonunu negatif yönde etkilemesi ve PO-R ve MPL sinyallerini inhibe etmesi ile önemli bir role sahiptir.107,108
Son yıllarda özellikle exon 2’de tanımlanan LN mutasyonları JA ileti sisteminin inhibisyonunu ortadan kaldırdığı için MPN nedeni olarak düşünülmektedir. LN mutasyonları nadirdir ancak daha sıklıkla lösemik transformasyon olan MPN olgularında daha sıklıkla görülmektedir (%13).109
IKZF Delesyon Mutasyonu
Ikaros Family Zinc Finger Protein (I ZF1) geni, normal hematopoezin gelişiminde fonksiyonel önemi olan Ikaros transkripsiyon faktörünü kodlayan 7p.12’de lokalize olan gendir. RNA aracılı I ZF1 eksikliğinin sitokin duyarlılığında ve p-STAT5 ekspresyonunda artışa neden olduğu gösterilmiş ve bu durumda I ZF1 kaybının MPN hastalarının bir kısmında lösemik transformasyonda önemli rol oynadığı söylenebilir.110 CASITAS B-Cell Lenfoma Mutasyonu
Casitas B-cell lenfoma (CBL) gen ailesi c-CBL, CBL-b ve CBL-c’yi içermektedir. CBL proteinleri ubiquitin ligaz aktivitesi olan multifonksiyonel proteinlerdir. Genellikle tirozin kinaz reseptörleri üzerinde inhibisyon yaparlar. CBL proteinlerinin birçok hedefi içerisinde JA reseptörleri de mevcuttur. c-CBL 11q.23.3’te yerleşmiştir ve mutasyonu birçok miyeloid malignitede tanımlanmıştır.
MPN’lerin kronik fazında c-CBL mutasyonları bazı PMF hastalarında düşük yüzdelerde (%6) bulunmuştur. Bununla birlikte bazı vakalarda hastalık progresyonunda JA 2V617F mutasyonunun kaybolduğu olgularda sonradan ortaya çıktığı gözlenmiştir.111-113
EZH2 Mutasyonu
EZH1 (The Polycomb Group Protein Enhancer of Zeste Homolog 1) ve EZH2 (The Polycomb Group Protein Enhancer of Zeste Homolog 2) proteinleri Policomb represiv kompleks 2 (PRC2)’ye aittirler. Proliferasyon, diferansiasyon, hücre kimliğinin devamı, yaşlanma ve esneklik gibi birçok hücre sürecinde rol alırlar. Ayrıca kromatin yapı düzenlenmesine de katkıda bulunurlar. Z 2 PRC2’nin iki adet alt bölümünü kodlar. Z 2 aşırı ekspresyonu prostat ve meme kanseri gibi birçok solid tümörde
21 görülür. Z 2 mutasyonu T’de görülmez ancak PV hastalarının %3’ünde ve PMF hastalarının %13’ünde görülmektedirler ve kötü prognoz ile ilişkilendirilirler.114
RAS Mutasyonları
Ras proteinleri, küçük guanozin-5-trifosfataz ailesine aittir.115,116
RAS genine ait üç onko enik mutasyon RAS, NRAS ve RAS mutasyonlarıdır. RAS ailesi mutasyonları arasında NRAS aktive edici mutasyonları, AML, MDS ve MPN’leri içeren hematopoetik malignitelerin yaklaşık %30’unda bulunmaktadır.117,118
NRAS/KRAS mutasyonları PV’da nadir görülmekle beraber PV’dan akut lösemiye transformasyonla ilişkisi olduğu gösterilmiştir.118
SOCS Mutasyonları
Sitokin sinyal supresör (SOCS) proteinleri JA ileti sistemi için önemli negatif regülatörlerdendir. SOCS kaybı sitokinler üzerinden sinyal artışlarına sebep olurlar. MPN hastalarında çok az görülmektedirler ve hastalık patogenezindeki rolleri ve önemi açıklığa kavuşmuş değildir.119
TET2 Mutasyonu
Ten- leven Translocation (T T) gen ailesi, T T onkogen ailesi üyesi 1 (T T1), T T onkogen ailesi üyesi 2 (T T2) ve T T onkogen ailesi üyesi 3’den (T T3) oluşmaktadır.120-122
T T2 geni, kromozom q2 ’te yerleşim göstermekle beraber metilsitozine hidroksil eklenmesini sağlayan hidroksilaz enzimini kodlar.120 TET2 mutasyonları DNA metilasyonunu arttırmakla beraber hidroksimetil sitozin seviyesinde azalmaya neden olur. T T2 mutasyonları, JA 2V617F veya MPL mutasyonlarından önce veya sonra ortaya çıkabilir.
TET2 mutasyonları miyeloid malignitelerde yaklaşık %15 oranında bulunmaktadır.123-127
T T2 mutasyon sıklığı PMF’de %17, T’de %11 ve PV’da %7-16 oranında bildirilmiştir. Bu mutasyonun tromboz oranını, lösemik transformasyonu veya tüm yaşam süresini etkilemediği bildirilmiştir. PMF hastalarında T T2 mutasyon durumunun yeni bir prognostik belirteç olmadığı düşünülmektedir. 128
ASXL1 Mutasyonu
ASXL1 (Additional sex comb-like 1) ASXL2 (Additional sex comb-like 2) ve ASXL 3 (Additional sex comb-like 3) ile birlikte OX genini baskılayan Asx (Drosophilia melanogaster additional sex combs) geni ile ilişkilidir. ASXL1 mutasyonları çerçeve kayması mutasyonları şeklinde olup genin 12. exonunda yer alırlar ve genellikle karboksi terminalinde P D domain kaybı ile kendilerini gösterirler. T ve
22 PV hastalarında görülme sıklığı %7’nin altında, PMF’de bu oran %19- 0 arasındadır. MPN’de görece sık görülen bir mutasyon olmasına karşın hematopoezdeki rolü net olarak anlaşılamamıştır.91,129
IDH1/IDH2 Mutasyonu
IDH1 (izositrat dehidrogenaz 1) ve IDH2 (izositrat dehidrogenaz 2) genleri izositrat dehidrogenaz 1 ve 2 enzimlerini kodlar. Bu enzimler izositratı alfaketogluterata dönüştüren NADP+ enzimleridir. ID 1 (R132S, R132G) ve ID 2 (R1 0,R172) ‘deki heterozigot mutasyonlar AML’de tanımlanmışlardır. Bununla birlikte düşük insidanslarda özellikle kronik faz T, PV ve PMF’de bulunmuşlardır. Bu durum göz önünde bulundurulunca bu mutasyonların lösemik transformasyon ile ilgili olduğu düşünülmekle beraber henüz yeterli çalışması bulunmamaktadır.130
CALR Mutasyonu
MPN hastalarında 2005 yılından sonra yapılan çalışmalarda T ve PMF’de hastaların yaklaşık %35- 0’ında JA 2 ve MPL mutasyonları negatif bulunmuştur. Bu hastaların patogenezine yönelik yapılan tüm ekzom sekanslama çalışmaları sonrasında Klampfl ve Nangalia’nın araştırma grupları tarafından 2013’de JA 2 negatif T ve PMF hastalarda calreticulin (CALR) somatik gen mutasyonu tanımlanmıştır.10,13 CALR geni 9 ekzondan oluşur ve 19p13.2 de lokalizedir.10
CALR şaperon aktivitesi ve kalsiyum homeostazında rol alan, hücre çoğalması, farklılaşması ve apopitozis gibi immüno enik hücre ölümü gibi birçok fonksiyonu olan yüksek derecede korunmuş bir endoplazmik retikulum ( R) proteinidir. R içinde ve dışında bulunarak lipid ve protein sentezi, Ca+2 depolanması, posttranslasyonel modifikasyon gibi hücre zarı, sitoplazma, nükleus ve ekstrasellüler matrikste birçok hücresel fonksiyona sahiptir.11,12
2013’de lampfl ve arkadaşları JA 2 ve MPL mutasyonu negatif olan MP hastalarında tüm ekzom sekanslama çalışması sonrası 36’dan farklı CALR somatik mutasyonu tanımladılar. Bu çalışmada JA 2 ve MPL (-) hastalarda T’de %67 , PMF’de %88 CALR mutasyonu olduğu gösterildi.10
Aynı yıl Nangalia ve arkadaşlarının yaptığı tüm ekzom sekanslama çalışmasında ise %70-8 oranında CALR somatik mutasyonu saptandı. Tüm bu çalışmalardaki mutasyonlar ekzon 9’da insersiyon ve delesyon şeklindeydi.10,13
2013’den günümüze ekzon 9’da 50’den farklı CALR mutasyonu tanımlanmıştır ve hepsi de frameshift mutasyon niteliğindedir.13
n sık saptanan ( yaklaşık %80) mutasyon CALR 52-bp delesyon p.L367fs*46 (Tip 1 CALR mutasyonu) ve 5-bp insersiyonu p.K385fs*47 (Tip 2 CALR mutasyonu) olarak saptanmıştır.10 (Şekil 3.) Bu iki