Various substituted forms of the triphenylmethyl (trityl) group continue to be used as site-selective protecting groups in the synthesis of amines and alcohols. X-ray structure determinations were examined both for confirmation of the structures, especially of N-tritylglycine in view of an earlier report that this compound exists as a non-zwitter-ion form.1 By an X-ray crystal structure determination, we were also able to correct an earlier literature2supposition regarding the structure of ditritylhydroxylamine.3 In our recent work, we showed the values of trityl and dimethoxytrityl for selective protection of sulfur and amine functions against the hydroxyl group in ambident nucleophiles.4 In the present study, the crystal structure of the title compound in Fig. 1 was determined by X-ray analysis.
After trityl bromide (71 mg, 0.22 mmol) was dissolved in chloroform (3 cm3), freshly distilled m-toluidine (236 mg, 0.22 mmol) was added. The mixture was stirred overnight at room temperature. It was then extracted between water (5 cm3) and more chloroform (5 cm3). The organic phase was dried with Na2SO4, filtered, and evaporated to give a brown crystalline compound, which was purified on an alumina column (20%
ethyl acetate: petrol) to give the title compound (50 mg, 64%). Since it crystallized in chloroform, the crystal structure of the title compound consists one chloroform molecule in an asymmetric unit.
The X-ray data were obtained on a Nonius Kappa CCD diffractomer, using an X-ray tube target with Mo and a graphite-monochromator [λ(Mo Kα) = 0.71073 Å] at 293 K. Data were collected to a maximum θvalue of 25.3˚ by ϕand ω scans. The structure was solved by direct methods using SIR92, x231 ANALYTICAL SCIENCES 2007, VOL. 23
X-ray Structure Analysis Online
Crystal and Molecular Structure of N-Trityl-m-aminophenol
Chloroform Solvate
ˆ. Ç
ELˆK,*
1C. C. E
RSANLI,*
2M. A
KKURT,
3,†and ˆ. D
EMˆRTAÍ*
4*1 Department of Physics, Faculty of Arts and Sciences, Cumhuriyet University, 06532 Sivas, Turkey
*2 Department of Physics, Sinop Faculty of Arts and Sciences, Ondokuz Mayıs University, Sinop, Turkey
*3 Department of Physics, Faculty of Arts and Sciences, Erciyes University, 38039 Kayseri, Turkey
*4 Department of Chemistry, Faculty of Arts and Sciences, Gaziosmanpa
s
a University, 60250 Tokat, Turkey
The title compound, (C26H23N)3·CHCl3, was synthesized by the reaction of trityl bromide with m-toluidine in chloroform and crystalized in the trigonal R3 space group (Z = 6) with the one molecule of chloroform, and with a = 20.765(5), b = 20.765(5), c = 24.924(5)Å, α = β= 90˚, γ= 120˚, V = 9307(4)Å3, Z = 6 and D
x = 1.250 Mg m–3. A least-squares refinement gave a residual index of R = 0.065 for 3540 observed reflections. Three phenyl rings of the title compound are at angles of 60.39(13), 74.10(14) and 70.17(13)˚ to the six-membered ring of the planar aminophenol group. The dihedral angles between three phenyl rings are 73.60(16), 87.80(15) and 56.52(15)˚.
(Received October 11, 2006; Accepted July 23, 2007; Published on web November 29, 2007)
† To whom correspondence should be addressed. E-mail: [email protected]
2007 © The Japan Society for Analytical Chemistry
Fig. 1 Chemical scheme of the title compound (C26H23N)3·CHCl3.
Table 1 Details of the crystal data, data collection, and structure refinement
Formula: (C26H23N)3·CHCl3
Formula weight = 1167.73 Crystal system: trigonal
Space group: R3 Z = 6 a = 20.765(5)Å = = 90˚ b = 20.765(5)Å = 120˚ c = 24.924(5)Å V = 9307(4)Å3 Dx = 1.250 Mg m–3
No. of reflections measured = 3540
No. of reflections used in refinement = 2802 [I > 2 (I)]
max = 68.3˚ with Mo Kα R = 0.065
( / )max = <0.001
(Δ )max = 0.43 e Å–3
(Δ )min = –0.52 e Å–3
Measurement: Nonius Kappa CCD diffractometer Program system: COLLECT
Structure determination: direct methods (SIR-97) Refinement: full matrix least squares (SHELXL-97)
CCDC 623639 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. α β γ σ θ Δ σ ρ ρ
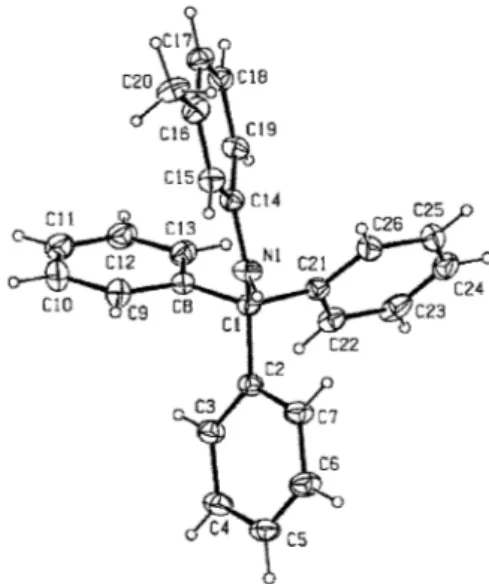
and refined by full-matrix least-squares procedures on F2, using the program SHELXL-97. An absorption correction was applied according to the multi-scan technique based on symmetry-related measurements. The non-hydrogen atoms were refined anisotropically, and all H atoms were positioned geometrically and allowed to ride on their parent atoms, with N–H = 0.86 and C–H = 0.93 or 0.98 Å, and with Uiso(H) = 1.2Ueq(C,N) or 1.5Ueq(Cmethyl) and refined to a final R (0.065). Details of the crystal data, data collection, and structure refinement are given in Table 1. The selected bond lengths and angles, and hydrogen-bond parameters are listed in Tables 2 and 3, respectively. An ORTEP-3 drawing of the title compound is shown in Fig. 2.
The title compound consists of a protecting group as trityl and o-methylaniline. The trityl group takes a propeller shape, as can be seen in Fig 1. The compound possesses normal geometric parameters. Three phenyl rings (A (C2–C7), B (C8–C13) and C (C21–C26)) in the title compound make dihedral angles of 60.39(13), 74.10(14) and 70.17(13)˚, respectively, with the six membered ring (D (C14–C19)) of the planar aminophenol group. The dihedral angles between three phenyl rings (A, B and C) are A/B = 73.60(16), A/C = 87.80(15) and B/C = 56.52(15)˚. The maximum deviations from the planarity of the N1 and C1 atoms through ring D (C14–C19) are –0.021(2) and 0.378(2)Å, respectively. Therefore, the N1 atom takes sp2 hybridization. In the intermolecular interaction between symmetry equivalent methyl groups of NH–Ph–CH3, because the van der Waals radius of the methyl group is generally 1.70 Å, the distance of the two carbon atoms of the methyl group should be larger than, or nearly equal to 3.40 Å. However, the distance
of C20·C20 (1–y, 1+x, z) in this structure is quite short, 2.771(4)Å. Moreover, the C16–C20 distance of 1.385(3)Å is too short as a methyl group. These abnormalities may indicate that C20 is an OH or NH2group, which has been interconnected by hydrogen bonds around three-fold rotation axis. However, this molecular structure was re-confirmed by the synthetic scheme, IR and NMR spectra. This may be because the title compound recrystalized with one solvent chloroform molecule in an asymmetric unit.
In the structure, there are no classical hydrogen bonds. Only one intra C–H·N type hydrogen bonding contact exists.
References
1. M. Canle, L. W. Clegg, I. Demirtas, M. R. J. Elsegood, and H. Maskill, J. Chem. Soc., Perkin Trans., 2000, 2, 85. 2. A. Mothwurf, Ber. Dtsch. Chem. Ges., 1904, 37, 3150. 3. M. Canle, L. W. Clegg, I. Demirtas, M. R. J. Elsegood, J.
Haider, H. Maskill, and P. C. Miatt, J. Chem. Soc., Perkin Trans., 2001, 2, 1742.
4. I. Demirtas, B. Büyükkıdan, and M. Elmastas, Turk. J. Chem., 2002, 26, 889.
x232 ANALYTICAL SCIENCES 2007, VOL. 23
Table 2 Geometric parameters (Å, ˚) for the title compound
Symmetry codes: (i) –y, x–y, z; (ii) –x+y, –x, z.
Table 3 Hydrogen-bond parameters (Å, ˚) for the title compound
D-H H...A D...A D-H...A
C26-H26...N1 0.93 2.42 2.790(4) 104.
Fig. 2 ORTEP-3 plot of the title compound, without the solvent molecule. Displacement ellipsoids for non-H atoms are drawn at the 20% probability level.