Downloaded from https://journals.lww.com/neurosurgery-quarterly by BhDMf5ePHKav1zEoum1tQfN4a+kJLhEZgbsIHo4XMi0hCywCX1AWnYQp/IlQrHD3tIQ5gQCIeyxFU0HdxzSR9vXTFnVX+PfRcL8ghidHdpm7540YOL1NUQ== on 01/10/2020 Downloadedfrom https://journals.lww.com/neurosurgery-quarterlyby BhDMf5ePHKav1zEoum1tQfN4a+kJLhEZgbsIHo4XMi0hCywCX1AWnYQp/IlQrHD3tIQ5gQCIeyxFU0HdxzSR9vXTFnVX+PfRcL8ghidHdpm7540YOL1NUQ==on 01/10/2020
Pseudopolyneuritic Form of Amyotrophic Lateral
Sclerosis With Foot Drop: Anatomic
and Electrophysiologic Study
Pınar Karakas¸, MD,* Filiz Koc¸, MD,
w
and Memduha G. Bozkır, PhD*
Abstract: The pseudopolyneuritic form of amyotrophic lateral sclerosis (ALS) is a subtype of ALS characterized by distal weakness of the lower limb and absence of Achilles tendon re-flex. Moreover, this form could be presented with foot drop. Therefore, the diagnosis of this form of ALS is important for clinicians because the clinical findings could indicate peripheral neuropathy. The aim of this study was to define the significance of the pseudopolyneuritic form of ALS with foot drop. We analyzed the clinical records of 138 (males, n = 83; females, n = 55) ALS patients who were admitted to the Department of Neurology between January 1995 and September 2011. Amyo-trophic Lateral Sclerosis Functional Rating Scale-Revised was used to quantify the patients. Onset complaints were in the upper extremity (n = 73), lower extremity (n = 33), bulbar involving (n = 25), dizziness (n = 3), unilateral paresthesia, and arm pain (n = 4). Among the patients with lower-limb onset, in 4 patients, the onset of lower-limb weakness was associated with distal dominance and absence of Achilles tendon reflex with foot drop. All these patients were male. In additional, 2 of the 4 patients are still alive, 1 of the patients died because of lym-phoma and the other died because of respiratory failure. This form of ALS had better survival than other phenotypes. Our findings underline the clinical importance and correct diagnosis for this form of ALS with foot drop.
Key Words: amyotrophic lateral sclerosis, foot drop,
pseudopolyneuritic form (Neurosurg Q 2013;23:289–293)
M
otor neuron disease (MND) or amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder characterized by progressive muscular paralysis reflecting the degeneration of motor neurons in the primary motor cortex, brainstem, and spinal cord.1–6This definition also holds for the common form of the disease, classical (Charcot’s) ALS. The incidence of sporadic ALS is re-ported to be between 1.5 and 2.7/100,000 population/y inEurope and North America. In the etiopathogenesis of ALS, several factors may be important including genetic factors, excitotoxicity, oxidative stress, mitochondrial dysfunction, neurofilament, and protein aggregation.4,7–11 The mean age of onset for the disease varies between 55 and 65 years, and males are affected more than females, with an M:F ratio of 1.5:1. Other syndromes related to this spectrum of disorders include progressive bulbar palsy, progressive muscular atrophy, primary lateral sclerosis, flail arm syndrome, and flail leg syndrome (pseudopolyneuritic form of ALS).
The pseudopolyneuritic form of ALS is charac-terized by distal weakness of the lower limb and absence of Achilles tendon reflex (ATR) at the onset of the disease.5,12,13 In additional, the patellar and upper limb tendon reflexes may show hyperreflexia.5,12,14
Fur-thermore, the prognosis of this form was reported to be significantly better than the other clinical phenotypes. Moreover, this form of ALS may be accompanied by foot drop because of effects on nervus peroneus. The common peroneal nerve is derived from the dorsal branches of the fourth and fifth lumbar and the first and second sacral ventral rami. It gives off superficial and deep peroneal nerves.15 Foot drop is a distressing problem because of the weakness of the dorsiflexor muscles of the foot. Although the most common cause of foot drop is per-oneal neuropathy, other causes including MND, lumbar plexopathies, trauma, myopathies, and L5 radiculopathy should also be taken into consideration.16–18 Therefore, evaluation of the pseudopolyneuritic form of ALS is im-portant for the clinicians because the clinical findings of this disease suggest peripheral neuropathy. Moreover, to date, information on the clinical features of this form has been inadequately studied. The aim of our study is, therefore, to describe the clinical profiles of 4 patients with the pseudopolyneuritic form of ALS, including foot drop, and to aid the clinician with a differential diagnosis.
MATERIALS AND METHODS
In this study, we enrolled 138 patients with ALS who were admitted to the Department of Neurology between January 1994 and September 2011. The demo-graphic characteristics and Amyotrophic Lateral Sclerosis Functional Rating Scale-Revised scores of our patients
From the Departments of *Anatomy; and wNeurology, Faculty of Medicine, C¸ukurova University, Adana, Turkey.
The authors declare no conflict of interest.
Reprints: Pınar Karakas¸, MD, Department of Anatomy, Faculty of Medicine, C¸ukurova University, Adana 01330, Turkey (e-mail: [email protected]).
were determined (Table 1). ALS was diagnosed by a senior neurologist (F.K.) depending on the neurologic and electrophysiologic examination and the Revised El Escorial criteria for ALS. All the patients were clinically positive for ALS according to the Revised El Escorial criteria. The mean age was 51.4 years (range, 38 to 76 y); the average age of the males was 51.5 ± 9.7 years (range, 38 to 76 y) and that of the females was 50.1 ± 8.6 years (range, 43 to 65 y).
The mean age at onset was 50.1 years (range, 37 to 73 y). Onset complaints were in the upper extremity (n = 73), lower extremity (n = 33), bulbar involving (n = 25), dizziness (n = 3), unilateral paresthesia, and arm pain (n = 4). Among the patients with lower-limb
onset, we found that 4 patients had onset of lower-limb weakness with distal dominance and absence of ATR with foot drop.
RESULTS
Case Reports
None of the 4 patients had any family history of ALS-like disorders. In additional, the superoxide dis-mutase (SOD1) gene mutation was negative in our pa-tients. The clinical features of the pseudopolyneuritic form of ALS cases are summarized in Table 2. Fur-thermore, cerebral and spinal magnetic resonance imag-ing (MRI) of the patients was found to be normal.
Case 1
A 52-year-old male was admitted to the neurology clinic with complaints of walking difficulty. This difficulty had begun 10 months ago. His medical history was not remarkable. On neurologic examination, there was mild weakness (4/5) in the extension and flexion of the left foot. However, he was unable to move his right foot (0/5). Atrophy was noted in the thenar and peroneal muscle groups. Fasciculation was observed in the extremity muscles. Deep tendon reflexes were absent in the lower extremities. Babinski signs were indifferent bilaterally. The patient had a steppage gait on the right side. There was no abnormality in any laboratory parameter, in-cluding complete blood count, routine biochemical anal-ysis, protein electrophoresis, Bence Jones protein, and CSF protein level. Cerebral and spinal MRI was normal. Electromyography of 4 limbs showed motor unit action potentials of long durations with fasciculation. Fibrilla-tion was observed in the musculus biceps brachii, m.flexor digitorum superficialis, m.abductor pollicis brevis,
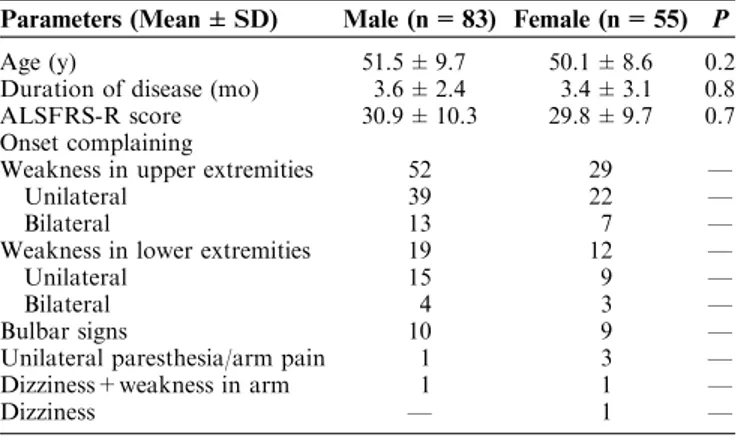
TABLE 1. Clinical Findings of Patients With Amyotrophic Lateral Sclerosis by Sex
Parameters (Mean ± SD) Male (n = 83) Female (n = 55) P
Age (y) 51.5 ± 9.7 50.1 ± 8.6 0.2
Duration of disease (mo) 3.6 ± 2.4 3.4 ± 3.1 0.8 ALSFRS-R score 30.9 ± 10.3 29.8 ± 9.7 0.7 Onset complaining
Weakness in upper extremities 52 29 —
Unilateral 39 22 —
Bilateral 13 7 —
Weakness in lower extremities 19 12 —
Unilateral 15 9 —
Bilateral 4 3 —
Bulbar signs 10 9 —
Unilateral paresthesia/arm pain 1 3 —
Dizziness+weakness in arm 1 1 —
Dizziness — 1 —
ALSFRS-R indicates the Amyotrophic Lateral Sclerosis Functional Rating Scale-Revised.
TABLE 2. Clinical Features of 4 Patients
Case 1 Case 2 Case 3 Case 4
Age (y) 52 38 58 70
Sex M M M M
Age of onset (y) 51 38 57 64
Disease duration (mo) 26 62 58 49
Onset complaint Weakness in the left lower limb
Weakness in the left lower limb
Weakness and numbness in the
left lower limb
Weakness in the right lower limb Distribution of weakness Right limb and upper
extremities
Right limb and upper extremities
Right limb and upper extremities
Left limb and upper extremities Deep tendon reflex at onset
Upper limb (right/left) Normal/normal Normal/normal Normal/normal Normal/normal Patellar (right/left) Normal/normal Normal/normal Normal/normal Normal/normal Achilles (right/left) Hypoactive/absent Absent/absent Absent/absent Absent/absent Bulbar symptom (initially/eventually) Absent/present Absent/present Absent/present Absent/present Upper motor neuron sign/through
the course)
Absent Present Absent Present
ALSFRS-R (initially) 40 38 40 —
ALSFRS-R (last) 4 16 20 6
Survival month Exitus-17 56 60 Exitus-36
SOD1 gene mutation Absent Absent Absent Absent
m.quadriceps femoris, m.tibialis anterior, m.extensor digitorum brevis, and m.abductor hallucis longus. In electroneurography, the left nervus fibularis was found to be inexitable and right fibular nerve conduction time was normal (49 m/s). The other motor and sensory con-duction times were normal. The patient was diagnosed with the pseudopolyneuritic form of ALS, and treatment with Riluzole tablet 100 mg/d was started. The patient died because of respiratory failure after 26 months.
Case 2
A 38-year-old male was admitted to the neurology clinic with complaints of gait disorder that started 8 months ago. He consulted a general hospital and was diagnosed with peroneal neuropathy. On neurologic ex-amination, there was mild weakness (4/5) in the extension and flexion of the right foot. However, he was unable to move dorsiflexion of his left foot (1/5). Atrophy was noted in the thenar and peroneal muscle groups. Fas-ciculation was observed in the extremity muscles. Deep tendon reflexes were brisk, except for the Achilles reflexes. Babinski signs were indifferent bilaterally. The patient had a steppage gait on the right side. Laboratory tests, including total blood count, routine biochemical tests, protein electrophoresis, Bence Jones protein, and CSF protein level were normal. Hormone profiles, including thyroid function tests, vitamin B12, and folic acid levels,
were normal. Cerebral and cervical, thoracal, and lum-bosacral MRI were normal. Electromyography showed neurogenic units and fasciculation and fibrillation in the upper and lower extremity muscles. Sensory and motor nerve conduction velocities were normal, except for the left fibular nerve. The left fibular nerve was inexitable. A clinical diagnosis of the pseudopolyneuritic form of ALS was made. Riluzole tablet 100 mg/d was started. The patient is still alive and the survival is now 62 months.
Case 3
A 58-year-old male was admitted to the neurology clinic with complaints of paresthesia and weakness in the left limb that started 12 months ago and showed pro-gression. Moreover, he had been diagnosed with lumbar herniated nucleus pulposus, and had been offered an operation. Neurologic examination indicated diminished gag reflex and slightly paretic plicas. Neck flexion was paretic (3/5). Tetraparesia was found in the distal muscle groups of upper extremities (4/5), proximal muscle groups (5/5), hip flexions (4/5), knee flexions, and extensions (4/5) and (3/5). In the right feet, plantar flexion was 4/5 and dorsiflexion was 1/5. Deep tendon reflexes were normoactive, except for the Achilles reflexes. ATRs were absent. The Babinski sign was indifferent bilaterally. Tongue, interosseal, tenar, hypotenar, forearm, arm, and peroneal group muscles were atrophic. Fasciculation was observed in the tongue and extremity muscles. Foot drop was present on the left side and steppage gait in the right lower limb. Laboratory tests including total blood count, routine biochemical tests, protein electrophoresis, Bence
Jones protein, and CSF protein level were normal. Cer-vical, thoracal, and lumbosacral MRI showed bulging disk at the C5-C6. Electromyography showed neurogenic units and fasciculation and fibrillation in the upper and lower extremity muscles. Sensory and motor nerve con-duction velocities were normal, except for the left fibular nerve. The left fibular nerve was inexitable. With the clinical and electrophysiologic findings, the patient was diagnosed with symptomatic MND and treatment with Riluzole tablet 100 mg/d was started and he was dis-charged. During the follow-up, the complaints pro-gressed. The patient is still alive and the survival is now 58 months.
Case 4
A 70-year-old man was admitted to the clinic with weakness in the right foot. He did not have a family history. The results of a physical examination were nor-mal. The results of a neurologic examination showed monoparesis and atrophy of the right foot (2/5) and fas-ciculation prominent in the upper and lower proximal muscle groups. Deep tendon reflexes were normoactive, but ATRs were absent. The Babinski sign was indifferent bilaterally. Hoffman-Tro¨mner and palmomental reflexes were bilaterally positive. Foot drop was present on the right side and steppage gait in the right lower limb. The results of standard laboratory analyses for total blood count, routine biochemical tests, protein electrophoresis, Bence Jones protein, and CSF protein level were normal. The results of abdominopelvic ultrasonography and spinal MRI did not show any pathologic findings. Elec-tromyography of 4 limbs showed motor unit action po-tentials of long durations with fasciculation. Fibrillation was observed in the m.rectus femoris, m tibialis anterior, and m gastrocnemius of the right lower limb. In electro-neurography, right n.fibularis were found inexitable and left fibular nerve conduction time was decreased (34 m/s). The other motor and sensory conduction times were normal. The patient was diagnosed with a pseudopoly-neuritic form of ALS. Riluzole (100 mg/d) and vitamin E (600 mg/d) were begun. Two years later, the patient died because of lymphoma.
DISCUSSION
ALS is the most common degenerative disease of the motor neuron system. Furthermore, as we mentioned before, 1 of the etiopathologic factors for ALS is genetic factors. Twenty percent of patients with autosomal dominant familial ALS and 2% of patients with sporadic ALS show mutations in the copper-zinc SOD1 gene.4,7 In our patients, the SOD1 gene mutation was found to be negative.
The pseudopolyneuritic form of ALS is a subtype of ALS characterized by distal weakness of the unilateral lower limb and absence of ATR at disease onset.5,12–14The pathology of the pseudopolyneuritic form of ALS is defined with myelinated fiber loss in the tractus corticospinalis of the thoracic and lumbar spinal cord segments.19,20
The pseudopolyneuritic form of ALS was first recognized by Pierre Marie and first described by Patrikios and it was known as the Marie-Patrikios form or the peroneal form of ALS.12,19,21 They described a syndrome of distal onset weakness and wasting of the lower limbs, which was asymmetric in onset, with absent lower limb tendon re-flexes, slow progression, and subtle or late upper motor neuron signs. In our 2 patients, there were no upper motor neuron signs in the beginning of the disease. However, 2 patients had upper motor signs, including pathologic reflexes (Hoffman-Tro¨mner sign) and brisk deep tendon reflexes in a later period.
This form was more common in women compared with other ALS forms. It showed an equal M:F ratio in a London population, whereas the ratio was 5:1 in a Melbourne study.19,22 However, our patients were men.
Identification of this form of ALS is important for clinicians because these clinical findings could suggest peripheral neuropathy.5Our 4 patients showed weakness of the distal lower limb and absence of ATR with foot drop at disease onset. Although the most frequent cause of foot drop is peroneal nerve neuropathy, other causes include myopathies, L5 radiculopathy, sciatic neuro-pathy, and MND.16–18 This condition involves the muscles of dorsiflexion (m.tibialis anterior, m.extensor hallucis longus, and m.extensor digitorum longus) and the nervus peroneus communis.17 The common peroneal
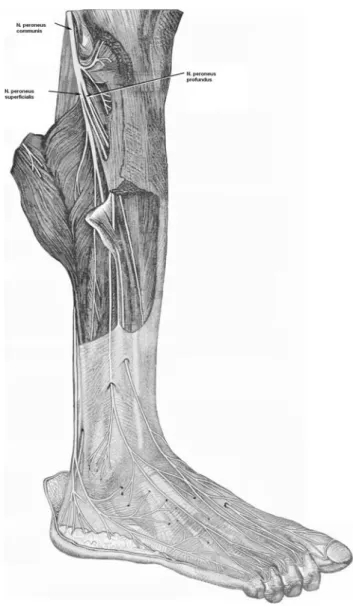
nerve is the lateral, smaller, and terminal branch of the sciatic nerve and divides into superficial and deep per-oneal nerves (Fig. 1). The deep perper-oneal nerve supplies m.tibialis anterior, m.extensor hallucis longus, m.extensor digitorum longus, and m.peroneus tertius. The superficial peroneal nerve supplies m.peroneus longus and m.per-oneus brevis.15,23 In individuals with foot drop, the foot
drops and the toes drag on the floor while walking. Because it is impossible to make the heel strike the ground first, the patient has a high stepping (steppage) gait, raising the foot as high as necessary to keep the toes from hitting the ground. In addition, the foot comes down suddenly, producing a clop.23
Some researchers, such as Wijesekera and col-leagues, have claimed that this form has a significantly better prognosis compared with bulbar and limb-onset ALS.19,24–26 The overall median survival of this form from the onset of symptoms was 69 months in a study including a London population, whereas it was 71 months in the Melbourne cases.19,22 We agree for this subtype’s having better prognosis that 2 of our cases are alive (survival 62 and 58 mo), 1 of them died from lymphoma and 1 case died as a result of respiratory failure.
As mentioned before, foot drop could suggest per-oneal neuropathy, but the appearance of fasciculations and having progression with other muscles weakness may provide a clue for MND and should be taken into con-sideration in the differential diagnosis. Furthermore, in electromyography, the presence of denervation potentials and giant units will also support the MND.
In summary, we define the pseudopolyneuritic form of ALS beginning with foot drop in 4 patients. This form
may be confused with some clinical conditions such as lumbar plexopathy, peroneal neuropathy, and distal myopathy because of the presence of foot drop. There-fore, a patient’s history, neurologic examination, and electrophysiologic evaluation should be assessed carefully for a differential diagnosis.
In conclusion, we believe that the pseudopolyneur-itic form of ALS that was presented in this paper has better prognosis and survival rates than the other sub-types of ALS. Moreover, this form could be present with foot drop. In this respect, it may be diagnosed as polyneuropathy. Thus, a detailed investigation including a neurologic examination and an electrophysiologic examination should be carried out for the correct diagnosis.
FIGURE 1. Anterolateral view of the right leg showing the common peroneal nerve, superficial, and deep peroneal branches.
REFERENCES
1. Kuleci S, Koc F, Hanta I. Profile of respiratory impairment in patients with amyotrophic lateral sclerosis. Neurosurg Q. 2010;4:288–291.
2. Koc F, Yerdelen D. Motor neuron disease and its association with non-Hodgkin’s lymphoma. Neurosciences. 2008;4:458–459. 3. Koc F, Paydas S, Yerdelen D, et al. Motor neuron disease
associated with multiple myeloma. Int J Neurosci. 2008;3:337–341. 4. Wijesekera LC, Leigh PN. Amyotrophic lateral sclerosis. Orphanet J
Rare Dis. 2009;4:3.
5. Kobayashi Z, Tsuchiya K, Arai T, et al. Pseudopolyneuritic form of ALS revisited: clinical and pathological heterogeneity. Neuropathol-ogy. 2010;30:372–380.
6. Nalini A, Thennarasu K, Gourie-Devi M, et al. Clinical character-istics and survival pattern of 1153 patients with ALS: experience over 30 years from India. J Neurol Sci. 2008;272:60–70.
7. Rosen DR, Siddique T, Patterson D, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62.
8. Shaw PJ. Molecular and cellular pathways of neurodegeneration in motor neuron disease. J Neurol Neurosurg Psychiatry. 2005;76: 1046–1057.
9. Ferrante RJ, Browne SE, Shinobu LA, et al. Evidence of increased oxidative damage in both sporadic and familial amyotrophic lateral sclerosis. J Neurochem. 1997;69:2064–2074.
10. Krasnianski A, Deschauer M, Neudecker S, et al. Mitochondrial changes in skeletal muscle in amyotrophic lateral sclerosis and other neurogenic atrophies. Brain. 2005;128:1870–1876.
11. Hirano A, Donnenfeld H, Sasaki S, et al. Fine structural observations of neurofilamentous changes in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 1984;43:461–470.
12. Patrikios JS. Contribution A l’Etude Des Formes Cliniques Et De l’ Anatomie Pathologique De La Sclerose Laterale Amyotrophique. Paris University. 1918.
13. Bonduelle M. Amyotrophic lateral sclerosis. In: Vinken PJ, Bruyn GW, eds. Handbook of Clinical Neurology 22. Amsterdam, North Holland: Churchill Livingstone; 1975:281–338.
14. Cappellari A, Ciammola A, Silani V. The pseudopolyneuritic form of amyotrophic lateral sclerosis (Patrikios disease). Electromyogr Clin Neurophysiol. 2008;48:75–81.
15. Williams PL, Bannister LH, Berry MM, et al. Gray’s Anatomy. 38th ed. Edinburgh: Churchill Livingstone; 1995:1286–1287.
16. Stewart JD. Foot drop: where, why and what to do? Pract Neurol. 2008;8:158–169.
17. Lavelle JM, McKeigue ME. Musculoskeletal dysfunction and drop foot: diagnosis and management using osteopathic manipulative medicine. J Am Osteopath Assoc. 2009;109:648–650.
18. Masakado Y, Kawakami M, Suzuki K, et al. Clinical neuro-physiology in the diagnosis of peroneal nerve palsy. Keio J Med. 2008;57:84–89.
19. Wijesekera LC, Mathers S, Talman P, et al. Natural history and clinical features of the flail arm and flail leg ALS variants. Neurology. 2009;72:1087–1094.
20. Terao S, Sobue G, Hashizume Y, et al. Disease-specific patterns of neuronal loss in the spinal ventral horn in amyotrophic lateral sclerosis, multiple system atrophy and X-linked reces-sive bulbospinal neuronopathy with special reference to the loss of small neurons in the intermediate zone. J Neurol. 1994;241: 196–203.
21. Hemmer R. On the peroneal form of amyotrophic lateral sclerosis. J Nervenarzt. 1955;26:400–401.
22. Talman P, Forbes A, Mathers S. Clinical phenotypes and natural progression for motor neuron disease: analysis from an Australian database. Amyotroph Lateral Scler. 2008;18:1–6.
23. Moore KL, Dalley AF. Clinically oriented anatomy. 4th ed. Lippincott Williams & Wilkins: Baltimore, Maryland; 1999:579–585. 24. Salemi G, Fierro B, Arcara A, et al. Amyotrophic lateral sclerosis in Palermo, Italy: an epidemiological study. Ital J Neurol Sci. 1989;10: 505–509.
25. Guidetti D, Bondavalli M, Sabadini R. Epidemiological survey of ALS in the province of Reggio Emilia, Italy: influence of environ-mental exposure to lead. Neuroepidemiology. 1996;15:301–312. 26. Mortara P, Bardelli D, Leone M, et al. Prognosis and clinical