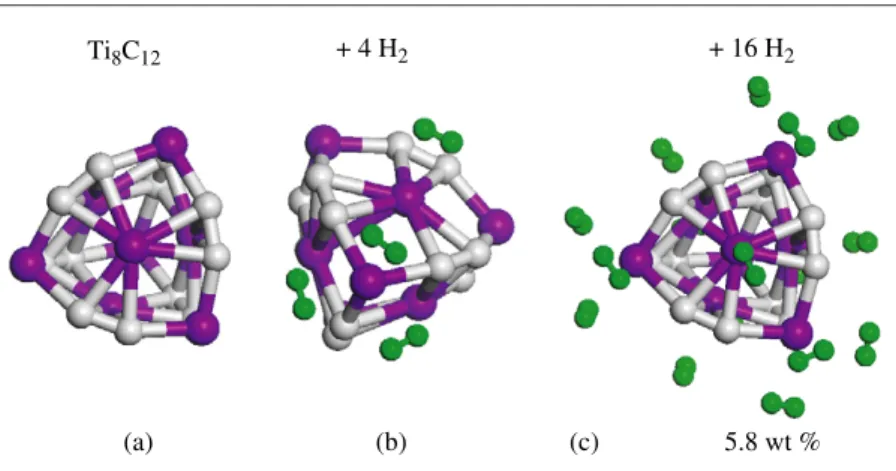
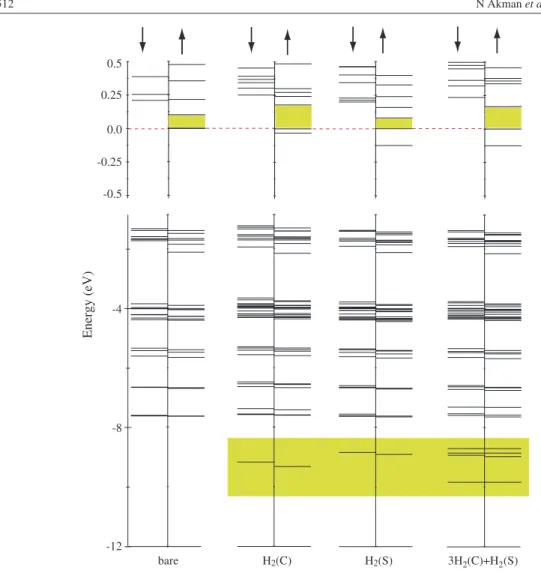
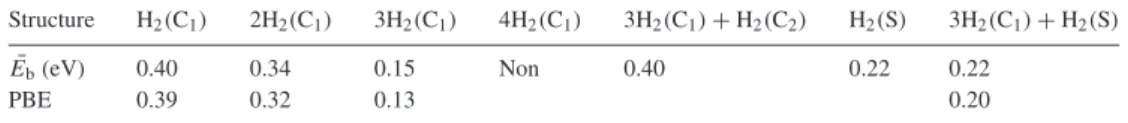
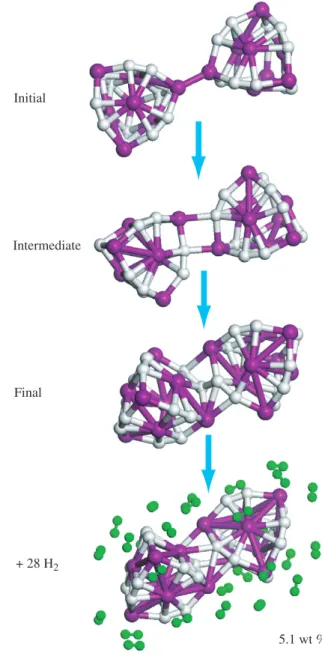
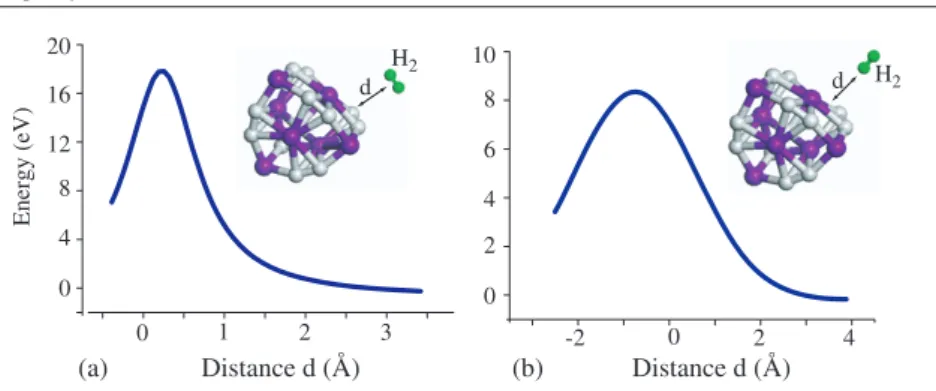
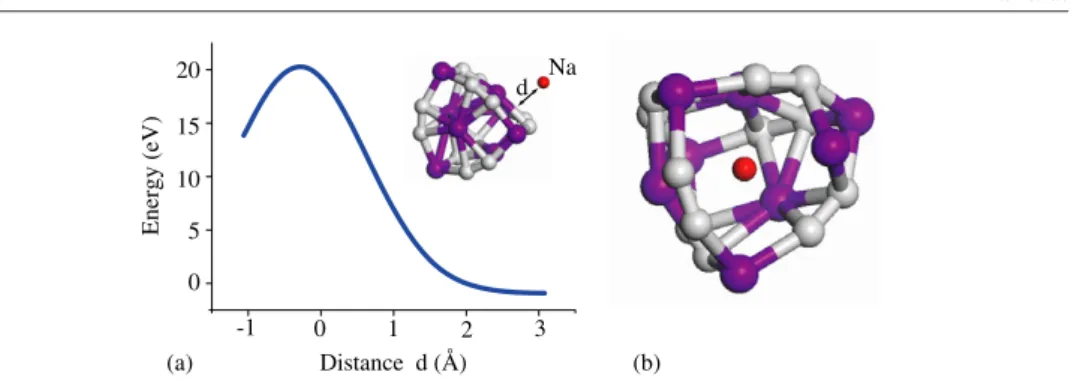
Hydrogen storage capacity of titanium met-cars
Tam metin
Şekil
Benzer Belgeler
Eradication of Helicobacter pylori and risk of peptic ulcers in patients starting long-term treatment with non-steroidal anti-inflammatory drugs: a randomised trial. Hawkey
Okul öncesi eğitim kurumuna devam eden 60-72 aylık çocukların annelerinin sahip oldukları sosyo-demografik özellikleri ve anne baba tutumlarının
Bulgular iki bölümden oluşmaktadır: Birinci bölümde, ölçek geliştirme ile ilgili bulgular ve ikinci bölümde örnekleme giren öğretmenlerin kişisel ve mesleki
Acil Sağlık Hizmetleri Yönetmeliği’ne göre triaj, çok sayıda hasta ve yaralının bulunduğu durumlarda, bunlardan öncelikli tedavi ve nakil edilmesi gerekenlerin tespiti
Bu kapsamda incelenen Birvan, Aşvan ve Meşeli cevherleşmeleri Yüksekova Karmaşığı'na ait derinlik kayaçlan tarafından kesilen Keban Metamorfıt- leri kontağı boyunca,
Mahınud Sadık Bey, hayatda ve mematda daima eyilik ve yük seklik ilham eden varlıklardandır, ölüleri, medh ve tebcile layık iseler, medh re tebcil etmekde, en
As the operation is done in liquid environment, the radiation impedance has to be modeled correctly and included in the mechanical side of the circuit model. It is important to
The major contribution of the paper can be stated as follows: In a neural network based learning task of distributed data, it is possible to obtain an accuracy almost as good as the
