Novel inhibitor design for histone demethylase 1 (lsd1) enzyme using molecular modeling
Tam metin
Şekil

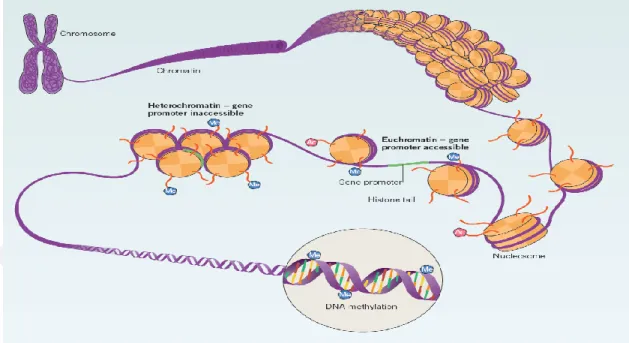



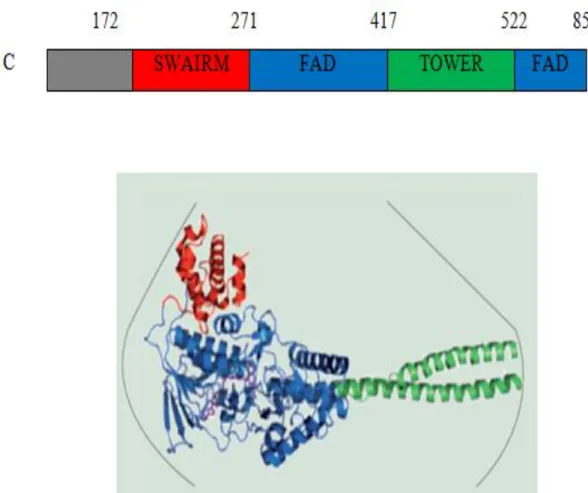

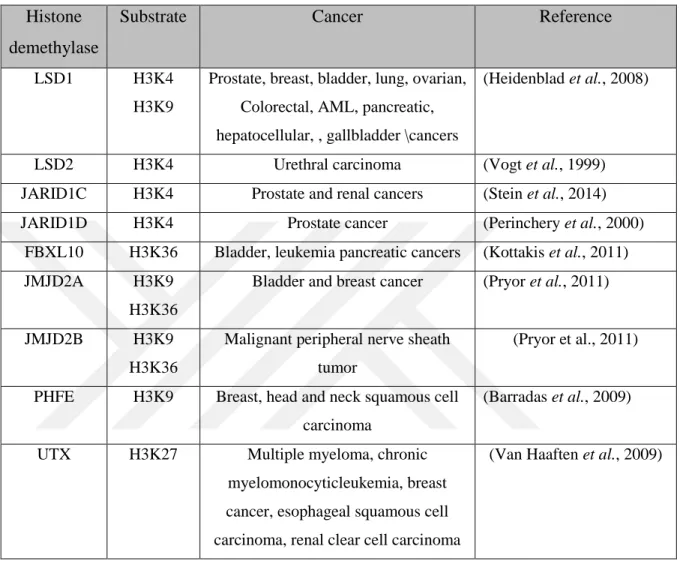
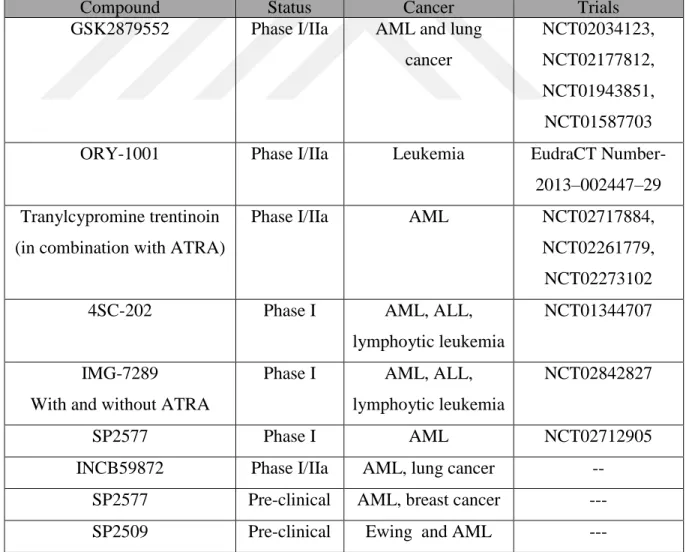

Benzer Belgeler
A total of 15 compounds obtained from the virtual screening (5 compounds for HDAC4; 3 compounds for HDAC5; 6 compounds for HDAC7; and 1 compound for HDAC9)
By looking at the solubility of an unknown substance in the solvents given below, an idea about the unknown substance can be obtained in three aspects (water, ether,
The aim of the present study is to identify the interaction patterns and binding affinity predictions of the selected PDE5 inhibitors (sildenafil, vardenafil and
Bir ta raftan yetmiş beş binlik bir kalabalık bu raya şehir ol demiş, semt semt bacalarını yükselten fabrikalar düdüklerini öttürerek mamur ol diye
In her works Herland and With Her In Ourland, Charlotte P. Gilman, aims to transfuse socialist and feminist ideologies. While being ahead of her time in certain aspects,
Değerlendirmeye katılan 40 hastanın 35'inde (%87,5) servikal lordoz açısı posterior tanjant yöntemine göre yaklaşık olarak normal kabul edilen 34°'nin altında kaldı..
Consequently, it is seen that almost all Turkish territorial waters in the Aegean Sea region and also a large part of high seas in this area are covered by
İkinci Dünya Savaşı sonrası insan hakları, demokrasi ve ifade hürriyeti gibi evrensel değerler temelinde bir bütünleşme sürecine giren Avrupa devletleri,
