Metal dicyanamides as efficient and robust water-oxidation catalysts
Tam metin
Şekil
![Figure 2. 1D chain structure of [Codca2]. Color code: Co= purple; O =red; C =gray; N=blue](https://thumb-eu.123doks.com/thumbv2/9libnet/5875813.121161/2.892.125.380.552.799/figure-chain-structure-codca-color-code-purple-gray.webp)
![Figure 3. XRD patterns of [Mdca2].](https://thumb-eu.123doks.com/thumbv2/9libnet/5875813.121161/3.892.453.823.97.332/figure-xrd-patterns-of-mdca.webp)
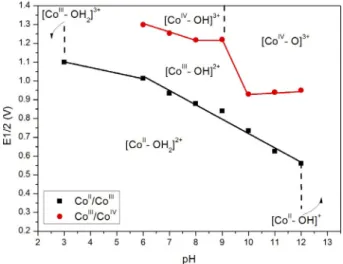
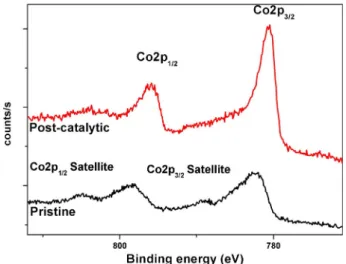

Benzer Belgeler
Bi rinci bölümde, “cinsiyet, mesleki deneyim, mezun oldu ğu okul, hizmetiçi eğitime katılma durumu, geçirdikleri teftiş sayısı” ile ilgili kişisel bilgiler
Yayımlanmamış yüksek lisans tezi, Ankara: Gazi Üniversitesi Sosyal Bilimler Enstitüsü, Sanat Tarihi Anabilim Dalı.. Eyüpsultan mezarlıklarında
This study aims to identify the impact of strategic management in the major charities in the Gaza Strip on transparency and relief of those affected in times of
Appendix 4.1 Table of the annual surface runoff (mcm) of the 10 rivers originating from Troodos Mountains.. Appendix 4.2 Table of the predicted annual surface runoff (mcm)
Its purpose is to demonstrate that research may benefit creative activity - not only in hindsight, whilst writing on completed output, but also as part of the creative process
青春痘粉刺護理 發佈日期: 2009/11/2 下午 05:22:48 更新日期: 2011-04-25 4:54 PM 一、何謂青春痘粉刺護理?
Bu sonuç, kamu sektörün- deki çalışanların özellikle çeşitli internet filtreleme ve izleme uygulamalarının olması ya da buna yönelik güçlü bir algının var olması
[r]
